Термодинаміка процесу адсорбції. Термодинаміка адсорбційних процесів Зміна термодинамічних функцій при адсорбції
Адсорбція як мимовільне концентрування молекул лежить на поверхні супроводжується зниженням ентропії системи. Оскільки критерієм мимовільності процесу є
∆Н - T · ∆S = ∆G< 0,
то адсорбція можлива лише за ∆Н< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. При підвищенні температури рівновага зміщується у бік ендотермічного процесу, тобто десорбції.
Адсорбція на поверхні твердого тіла
1. Мономолекулярна адсорбція.
По теорії Ленгмюра молекули адсорбтиву взаємодіють із поверхнею адсорбенту, створюючи у результаті мономолекулярний шар. В цьому випадку ступінь заповнення () поверхні адсорбованим речовиною при адсорбції з газової фази
з рідини
де К - константа рівноваги (константа адсорбції);
р - парціальний тиск газу, що адсорбується;
с - концентрація речовини, що адсорбується.
Залежність від р (або с) представлена графіком (ізотерма адсорбції, Т = const) на рис. 1.3.

Рис. 1.3. Ступінь заповнення поверхні речовиною, що адсорбується.
При малих концентраціях та парціальному тиску адсорбція пропорційна концентрації або парціальному тиску:
р<< 1, β ≈ К· р абос<< 1, β ≈ К· з, тобто. початкова ділянка ізотерми приблизно лінійна, причому tg α = К(tg α визначають по нахилу кривої при р (або с) → 0: або ).
Якщо кількість молей адсорбованої речовини на 1 г адсорбенту; - максимально можливу кількість молей адсорбованої речовини на 1 г адсорбенту ("ємність моношару"), то
Підставляючи β у рівняння (1.3) (для випадку адсорбції з газової фази концентрацію зу рівняннях слід замінити на тиск р), отримуємо:
 (1.6)
(1.6)
Оскільки і К у цій парі адсорбент-адсорбтив є константами (при T=const), то залежно можна знайти і До(Рис. 1.4).

Рис. 1.4. Графічне вирішення рівняння адсорбції
одержують шляхом екстраполяції експериментальної лінійної залежності до () = 0; і, тому що , то , .
Величину можна використовувати для визначення питомої поверхні адсорбенту УД (м 2 на 1 г адсорбенту), якщо відома площа ω, займана на поверхні однією молекулою адсорбтиву (визначається з розмірів молекули):
УД = · ω · Nа, (1.7)
де Nа – число Авогадро (Nа = 6,02 · 1023).
У свою чергу, відому величину УД можна використовувати для розрахунку або будь-якої речовини з його адсорбції на даному адсорбенті.
2. Полімолекулярна адсорбція.
Рівняння (1.5) визначає криву з насиченням, тобто. при
р (або с) → ∞ прагне граничного значення, що дорівнює (рис. 1.5,а).

Рис.1.5. Ізотерми адсорбції:
а - адсорбція з насиченням; б – полімолекулярна адсорбція
Однак у деяких випадках ізотерми адсорбції виглядають як показано на рис. 1.5, б, тобто. не досягає межі навіть за високих р (або с).
Залежність типу показаної на рис. 1.5 б відповідають полімолекулярної адсорбції. Як правило, такі ізотерми характерні для речовин із сильними міжмолекулярними взаємодіями (наприклад, для води). Коли центри адсорбції на поверхні адсорбенту зайняті (мономолекулярний шар насичений), посадка наступних молекул адсорбату відбувається за рахунок міжмолекулярних взаємодій з уже адсорбованими молекулами (рис.1.6). Теплота такої адсорбції близька за абсолютною величиною, але протилежна за знаком теплоти випаровування відповідної рідини (подумайте, чому).

Рис.1.6. Схема адсорбції:
а – мономолекулярна адсорбція; б - полімолекулярна адсорбція
У міру наближення рдо тиску насиченої пари адсорбируемого речовини воно починає конденсуватися на поверхні адсорбенту, в результаті швидко зростає зі зростанням р.
Термодинаміка адсорбційних процесів.
| Найменування параметру | Значення |
| Тема статті: | Термодинаміка адсорбційних процесів. |
| Рубрика (тематична категорія) | Освіта |
Основні визначення та способи класифікації адсорбційних процесів.
Адсорбція відноситься до явищ, що відбуваються внаслідок мимовільного зменшення поверхневої енергії.
Адсорбція- процес мимовільного оборотного або незворотного перерозподілу компонентів гетерогенної системи між поверхневим шаром та обсягом гомогенної фази.
У багатокомпонентних системах поверхневий шар переважно переходить компонент, який сильніше знижує міжфазне натяг. У однокомпонентних системах для формування поверхневого шару відбувається зміна його структури (певна орієнтація атомів і молекул, поляризація), зване автоадсорбцією.
Більш щільну фазу, на якій локалізовані адсорбційні взаємодії, називають адсорбентом. Речовина, що перерозподіляється між обсягом гомогенної фази і поверхневим шаром, позначають терміном адсорбатʼʼ.
У ряді випадків процес адсорбції є оборотним. В цьому випадку за певних умов частина адсорбованих молекул в результаті молекулярно-кінетичних явищ може перейти з поверхневого шару в об'єм фази. Процес, зворотний адсорбції, називають десорбцією.
Методи класифікації адсорбційних процесів.
Класифікація адсорбційних процесів за агрегатним станом взаємодіючих фаз.Враховуючи залежність отагрегатного стану суміжних фаз розрізняють такі типи адсорбційних процесів:
Адсорбція газів на жорстких адсорбентах;
Адсорбція розчинених речовин на межах розділу «тверде тіло – рідина» і «рідина – рідина»;
Адсорбція поверхнево-активних речовин на межі розділу «рідина – газ».
Класифікація адсорбційних процесів за механізмом взаємодії адсорбенту та адсорбату.Адсорбцію можна розглядати як взаємодію молекул адсорбату з активними центрами адсорбенту. За механізмом їхньої взаємодії поділяють такі види адсорбції:
1) фізична (молекулярна) адсорбція– взаємодія між молекулами адсорбату та адсорбенту здійснюється за рахунок сил Ван-дер-Ваальса, водневих зв'язків (без протікання хімічних реакцій);
2) хімічна адсорбція (хемосорбція)- Приєднання молекул адсорбату до активних центрів адсорбенту відбувається в результаті протікання хімічних реакцій різних типів (за винятком реакцій іонного обміну);
3) іонообмінна адсорбція (іонний обмін) - перерозподіл речовини адсорбату між розчином і твердою фазою (іонітом) за механізмом реакцій іонного обміну.
Для кількісного опису адсорбційних процесів застосовують дві величини.
1) Абсолютна адсорбція- Кількість (моль) або маса (кг) адсорбату на одиницю площі поверхні або маси адсорбенту. Позначення – А; розмірність: моль / м 2, моль / кг, кг / м 2, кг / кг.
2) Гіббсівська (надлишкова) адсорбція- Надлишок речовини адсорбату в поверхневому шарі певної товщини в порівнянні з його кількістю в обсязі гомогенної фази, віднесений до одиниці площі поверхні або маси адсорбенту. Позначення – Р; розмірність: моль / м 2, моль / кг.
Зв'язок між абсолютною та надмірною адсорбцією можна проілюструвати за допомогою рівняння:
Г = А - с * h (3.1)
де с - рівноважна концентрація речовини в об'ємі фази, моль/м 3;
h - товщина поверхневого шару, що умовно приймається рівною 10 -9 м.
У багатокомпонентних гетерогенних системах при перерозподілі того чи іншого компонента між обсягом гомогенної фази та поверхневим шаром справедливе рівняння для надлишкової внутрішньої енергії поверхні:
U = T * S + s * s + Sm i * n i (3.2)
Привівши всі члени рівняння до одиниці площі міжфазної поверхні, отримаємо:
U s = T * S s + s + Sm i * Г i (3.3)
де Г i = n i / s - надлишок i-го компонента в поверхневому шарі, тобто гіббсівська адсорбція.
Для однокомпонентної системи рівняння (3.3) набуде вигляду:
G s = s + m * Г (3.4)
де G s = U s - T * S s - Енергія Гіббса поверхні або робота створення одиниці площі поверхні;
m * Г – ущільнення речовини речовини, що адсорбується в поверхневому шарі.
Виходячи з рівняння (3.4) можна зробити висновок про те, що при адсорбції робота зі створення міжфазної поверхні складається з роботи утворення поверхні (розриву когезійних зв'язків в обсязі фази адсорбату) та ущільнення речовини у поверхневому шарі.
У стані динамічної рівноваги між адсорбентом і адсорбатом зміна енергії Гіббсу гетерогенної системи ΔG = 0, термодинаміка процесу адсорбції описується рівнянням, що отримало назву фундаментальне адсорбційне рівняння Гіббса:
Ds = SГ i * dm i (3.5)
Це рівняння є універсальним, оскільки справедливо для всіх типів адсорбційних процесів.
Окремі випадки адсорбційного рівняння Гіббса.
1) Адсорбція із розчинів.
Для хімічного потенціалу i-го компонента системи при протіканні адсорбції на межах розділу «рідина – твердий адсорбент» і «рідина – газ» справедливі рівняння:
m i = m i 0 + R*T*ln a i (3.6)
dm i = R * T * d ln a i (3.7)
де m i 0 - Хімічний потенціал i -го компонента системи за стандартних умов;
a i – активність i-го компонента системи за стандартних умов.
Виходячи з цього, адсорбційне рівняння Гіббса набуде вигляду:
Г i = - a i / R * T * (ds / da i) (3.8)
Для розчинів неелектролітів приймаємо a i = с i тоді:
Г i = - з / R * T * (ds / dс) (3.9)
Для розчинів електролітів:
Г i = - з ± n / R * T * (ds / dс ± n) (3.10)
де з ± – середня іонна концентрація розчину;
n – стехіометричний коефіцієнт.
2) Адсорбція речовин із газової фази.
Відповідно до рівняння Менделєєва-Клайперона:
Р = с * R * T (3.11)
У зв'язку з цим рівняння Гіббса для адсорбції газів на твердих адсорбентах записують у наступній формі:
Г i = - Р/R*T* (ds/dР) (3.12)
На практиці адсорбційне рівняння Гіббса дозволяє за даними вимірювання поверхневого натягу при різних значеннях концентрації рідини або рівноважного тиску газу розрахувати величину адсорбції речовин у міжфазному шарі, для якого визначено поверхневий натяг.
Термодинаміка адсорбційних процесів. - Поняття та види. Класифікація та особливості категорії "Термодинаміка адсорбційних процесів." 2017, 2018.
Поточна сторінка: 6 (всього книга 19 сторінок) [доступний уривок для читання: 13 сторінок]
Шрифт:
100% +
34. Природа адсорбційних сил
Взаємодія між молекулами адсорбтиву із поверхнею адсорбенту при т.з. фізична адсорбція може бути обумовлена різними причинами. Тоді потенціал, який обумовлює взаємодію однієї молекули адсорбенту з одним атомом неполярного адсорбтиву, можна виразити так:
θ = −Сr 6 +Br 12 ,
де r - Відстань між центрами частинок; С – константа дисперсійного тяжіння; В – константа, яка характеризує енергію сил відштовхування.
Цілком очевидно, що на порівняно віддалених відстанях мають переважати сили тяжіння, а на відстані близьких – сили відштовхування. Також на певних відстанях ці сили мають бути рівними, що відповідатиме мінімуму вільної енергії. Але важливо відзначити, що при адсорбції дисперсійні сили діють одночасно між кожною неполярною частинкою.
Оскільки енергія взаємодії частинок може швидко зменшуватися з відстанню, то визначення потенціалу адсорбційних сил досить провести підсумовування на найближчих атомах адсорбенту. Важливим є те, що при адсорбції складних неполярних молекул потенційну енергію можна приблизно підрахувати як суму всіх потенційних енергій адсорбції ланок молекули.
Якщо адсорбент складається з іонів, то до дії вже відомих дисперсійних сил може додаватися дія індукційних сил тяжіння диполів які індуковані в молекулах адсорбтиву електричним полем, яке, у свою чергу, створюється іонами решітки адсорбенту.
При такій взаємодії частка індукційних сил в адсорбційній взаємодії може бути пропорційна поляризуемості молекули адсорбтиву та квадрату напруженості поля на цій поверхні адсорбенту.
Якщо ж на полярному адсорбенті відбувається адсорбція полярних молекул адсорбтиву, то диполі у разі поляризують атоми адсорбенту, т. е. хіба що індукують у яких електричні моменти. Внаслідок такого впливу індукційна взаємодія додається до дисперсійної.
Сама індукційна взаємодія зазвичай мала і в залежності від диполя молекули адсорбтиву і поляризується адсорбенту може досягати великих значень. Якщо молекули адсорбуються на адсорбенті, який має на поверхні іони або диполі, виникає т.з. взаємодія іонів чи диполів адсорбтиву з електростатичним полем самого адсорбенту.
При цьому молекули адсорбтиву можуть навіть орієнтуватися в полі адсорбенту, при цьому відбувається орієнтаційна кулонівська взаємодія. Зазвичай буває, що енергії індукційної та орієнтаційної взаємодії менше енергії дисперсійної взаємодії, і тому сприймається, що енергія міжмолекулярного тяжіння визначається енергією дисперсійного тяжіння.
Також причиною адсорбції може бути утворення водневого зв'язку. Зв'язок такого типу може виникати при адсорбції на адсорбентах, які містять на поверхні гідроксильні групи таких молекул, як молекули води, спиртів, аміаку та амінів. При утворенні водневого зв'язку енергія взаємодії адсорбтиву з адсорбентом може бути досить великою, і теплота, що виділяється при такій адсорбції, значно більша за теплоту адсорбції речовин, які подібні за формою та розміром молекул, але не утворюють водневого зв'язку.
Важливо відзначити, що, знаючи термодинамічний опис поверхневого шару на межі «адсорбент – адсорбтив», його будову, природу різних видів сил, динаміку процесу можна переходити до вивчення більш складних процесів адсорбції.
35. Адсорбція як спонтанне концентрування на поверхні розділу фаз речовин, що знижують міжфазний натяг
Поверхнево-активні речовини поділяються на великі групи: активні та інактивніречовини.
Поверхнево-активні речовини здатні накопичуватися в поверхневому шарі, і при цьому відбувається позитивна адсорбція Г > 0.
Такі види речовин повинні володіти поверхневим натягом, яке, у свою чергу, повинно бути менше поверхневого натягу розчинника, або в іншому випадку накопичення речовини в поверхневому шарі буде невигідно, і повинні володіти порівняно малою розчинністю. При досить добрій розчинністю молекули поверхнево-активних речовин прагнуть піти з поверхні в глиб розчину. Отже, поверхнево-активні речовини переважно виштовхуватимуться з обсягу рідини на поверхню.
Але при накопиченні речовин на межі розчину в молекулах цих речовин, які слабо взаємодіють один з одним, міжмолекулярна взаємодія в поверхневому шарі зменшуватиметься, а поверхневий натяг падатиме.
Поверхнево-активними речовинамищодо водного шару є багато видів органічних сполук, жирні кислоти з досить великим вуглеводневим радикалом, солі цих кислот (мила), сульфокислоти та їх солі, а також різні види спиртів та амінів. Характерною особливістю більшості молекул є їхня дифільність: молекула складається з двох частин полярної групи та неполярного вуглеводневого радикала. Має значний дипольний момент і добре гідратує полярна група може обумовлювати спорідненість поверхнево-активної речовини до водного середовища. Але вуглеводневий радикал є причиною, що знижує розчинність цих сполук.
Поверхнево-інактивні речовини ПАР- ці види речовини, що прагнуть піти з поверхні рідини в її об'єм, в результаті відбувається т.з. негативна адсорбція Г < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Поверхнево-інактивними речовинамищодо води є багато неорганічних електролітів: кислоти, луги, солі. Молекули поверхнево-інактивних речовин не мають гідрофобної частини та можуть розпадатися у воді на добре гідратуючі іони.
прикладамиповерхнево-інактивних речовин є деякі органічні сполуки, у яких неполярна частина молекули відсутня або дуже мала. До таких речовин можна віднести мурашину, амінооцтову кислоти.
У неводних розчинниках неорганічні електроліти також здатні підвищувати поверхневий натяг, причому це залежить від розчинника.
Наприклад, при введенні йодиду натрію в метанол сильно підвищується поверхневий натяг, для етанолу поверхневе натяг більше приблизно в 2 рази. Поверхнева активність речовин може залежати не тільки від природи речовини, але також властивостей розчинника. Якщо будь-який розчинник має великий поверхневий натяг, то дана розчинена речовина може проявляти значну поверхневу активність.
36. Теорії адсорбції
Розглянемо найпоширеніші теорії адсорбції, що описують окремі види адсорбції на поверхні розділу "тверде тіло - газ" або "тверде тіло - розчин".
Теорія мономолекулярної адсорбції І. Ленгмюр.
1. Адсорбція є локалізованою та викликається силами, близькими до хімічних.
2. Адсорбція відбувається лише на активних центрах – виступах або западинах на поверхні адсорбенту, що характеризуються наявністю вільних валентностей. Активні центри вважаються незалежними та тотожними.
3. Кожен активний центр здатний взаємодіяти лише з однією молекулою адсорбату; на поверхні може утворитися лише один шар адсорбованих молекул.
4. Процес адсорбції є оборотним та рівноважним; адсорбована молекула утримується активним центром якийсь час, після чого десорбується; через деякий час встановлюється динамічна рівновага.
Максимально можлива величина адсорбції Гпро досягається за умови, що це активні центри зайняті молекулами адсорбату. Рівняння ізотерми мономолекулярної адсорбції, що зв'язує величину адсорбції Гз концентрацією адсорбату З, має вигляд:
де b– постійна для цієї пари «адсорбент – адсорбат» величина (ставлення констант швидкостей десорбції та адсорбції), чисельно рівна концентрації адсорбату, коли він зайнята половина активних центрів.

Графік ізотерми адсорбції Ленгмюра наведено малюнку 2. Константу bвизначимо графічно, провівши дотичну до ізотерми адсорбції в точці З= 0. При описі процесу адсорбції газів рівняння концентрація може бути замінена пропорційною величиною парціального тиску. Теорія мономолекулярної адсорбції І. Ленгмюра застосовна для опису процесів адсорбції газів та розчинених речовин при невеликих тисках (концентраціях) адсорбату.
Теорія полімолекулярної адсорбції Поляніописує s-подібні ізотерми адсорбції, форма яких свідчить про можливу взаємодію адсорбованих молекул з адсорбатом.
1. Адсорбція спричинена фізичними силами.
2. Поверхня адсорбенту однорідна, немає активних центрів; адсорбційні сили утворюють безперервне силове поле поблизу поверхні адсорбенту.
3. Адсорбційні сили діють на відстані, більшій за розмір молекули адсорбату, тобто у поверхні адсорбенту існує певний адсорбційний обсяг, який при адсорбції заповнюється молекулами адсорбату.
4. Притягнення молекули адсорбату поверхнею адсорбенту залежить від наявності в адсорбційному обсязі інших молекул, унаслідок чого можлива полимолекулярная адсорбція.
5. Адсорбційні сили не залежать від температури, і, отже, із зміною температури адсорбційний об'єм не змінюється.
Рівняння Фрейндліха.Поверхня адсорбенту неоднорідна, між адсорбованими частинками відбувається взаємодія, активні центри є повністю незалежними друг від друга. Г. Фрейндліх припустив, що кількість молей адсорбованого газу або розчиненої речовини, що припадає на одиницю маси адсорбенту (т.з. х/m), має бути пропорційно рівноважному тиску (для газу) або рівноважної концентрації (для речовин, що адсорбуються з розчину) адсорбенту, зведеної в деякий ступінь, який завжди менший за одиницю:
x / m = aP n; x / m = aC n.
Показники ступеня nта коефіцієнт пропорційності авизначаються експериментально.
37. Термодинаміка процесу адсорбції. Рівняння адсорбції Гіббса
Для вивчення явища адсорбції на межі «розчин – газ» необхідно встановити зв'язок між надлишком адсорбованої речовини у шарі на поверхні ( Г), концентрацією ПАР у розчині ( з) та поверхневим натягом ( σ ) на межі розділу фаз "розчин - газ". Доцільніше розглядати явища з термодинамічних позицій та пов'язувати адсорбцію розчиненої речовини із зміною вільної енергії поверхні або її поверхневого натягу. Цей зв'язок вивів В. Гіббсв 1876 р, яка отримала назву «Рівняння адсорбції Гіббса»:
Г = – з / RT x dσ/dc.
Ще можна уявити рівняння Гіббса,засноване на термодинаміці, з використанням ізобарно-ізотермічного потенціалу G, хімічних потенціалів μ 1і μ 2 ,а також із використанням n 1 і n 2 числом молей компонентів. Проаналізувавши його з урахуванням ентропії S, обсягу Vта тиску P, можна записати наступне рівняння:
dG=- SdT+VdP+σds+ μ 1 d n 1 + μ 2 dn 2 .
Прирівняємо його до нуля, і з урахуванням постійної температури та тиску воно спрощується в рівняння виду:
sd σ + n 1 d μ 1 + n 2 d μ 1 = 0.
З огляду на те, що з розбавлених розчинів хімічний потенціал другого компонента виражається так:
μ 2 = μ 2 0 +RT ln c,
а з огляду на те, що температура постійна
dμ 2 =RTdnc,
підставляючи це рівняння в
![]()
отримуємо шукане рівняння адсорбції Гіббса. Виходячи з рівняння, можна помітити, що якщо поверхневий натяг σ збільшується з концентрацією з, то концентрація розчиненої речовини на поверхневому шарі менша, ніж в об'ємі розчину (т. н. негативна адсорбція), і якщо поверхневий натяг σ зменшується із збільшенням концентрації з, тоді концентрація в шарі більша, ніж в обсязі (позитивна адсорбція), і, нарешті, якщо σ не залежить від зто концентрація речовини в шарі на поверхні та в обсязі однакова. Рівняння Гіббса виведено з використанням термодинаміки. Практично перевірити це рівняння складно, що пов'язано зі складністю визначення концентрації розчиненої речовини в слоєні поверхні. Досвідченим шляхом Б. Мак-Бен встановив, що з поверхні розчину за допомогою приладу зрізався тонкий шар рідини. Подальше визначення всіх параметрів рівняння Гіббса показало, що експериментально знайдені значення адсорбції в межах помилки досвіду співпадали зі значеннями, які обчислювали за рівнянням Гіббса. Через однорідність і гладкість поверхні будь-якої рідини при вивченні адсорбції на її поверхні абсолютно непридатні звичайні уявлення про активні центри. При критичній температурі зникає різницю між межами, поверхневе натяг, як правило, стає рівним нулю. Адсорбція газів і пар має настільки велике практичне застосування, що в літературі, особливо в технічній, можна зустріти це поняття, яке застосовують лише по відношенню до процесів на поверхні твердих тіл.
Це поняття, як і найбільш загальні закономірності адсорбції, як розглянуте рівняння Гіббса, застосовується до всіх меж розділу фаз. Користуючись рівнянням Гіббса і всіма положеннями, що випливають з нього, визначивши величину Г, можна побудувати ізотерму адсорбції.
38. Особливості адсорбції на мікропористих матеріалах. Потенційна теорія Поляні. Адсорбційний потенціал
Теорія Полянірозглядає нелокалізовану фізичну адсорбцію, яка безпосередньо зумовлена вандерваальсовими силами між адсорбентом та адсорбатом (це можна вважати першим положенням). Другим положенням цієї теорії є уявлення про силове (або потенційне) поле адсорбенту, яке поширюється на значну відстань від поверхні; шар адсорбції, який виникає у цьому полі, полімолекулярний. Якщо розглядати адсорбцію газів, тоді густина цього шару зменшується за певною нормаллю від поверхні. Якщо розглядати адсорбцію пари, тоді на поверхні утворюється рідкий шар певної товщини. Поля теоретично Поляні розглядають як ряд еквіпотенційних поверхонь, кожна поверхня відповідає певному значенню потенціалу ε , причому кожна наступна поверхня буде меншою, ніж попередня. Кожна така поверхня у просторі вирізує шари певного обсягу, позначеного як v i. Завданням теорії Поляні є знаходження переходу від звичайних координат ізотерми. x, p) до параметрів поля ε iі v iз подальшим встановленням зв'язку між цими основними параметрами. Перша частина задачі, яку заклав Поляні, досить складна, і в більшості випадків не може мати певних рішень, але для адсорбції парів ця частина завдання вирішується в першому наближенні дуже просто. Для рідкого адсорбційного шару заповнена частина обсягу дорівнюватиме:
v i = х(М/d),
де d- Щільність речовини в рідкому стані.
У своїй теорії M. Поляні запроваджує ще одне положення про відсутність т.з. екранування поля в процесі адсорбції, величина ε в даній теорії простору є величиною постійної (щось на кшталт гравітаційного потенціалу) незалежно від того, чи існують певні молекули адсорбату між даною точкою і твердою поверхнею або весь простір є вільним. Поляні запроваджує поняття адсорбційного потенціалу ε , який являє собою ізотермічну роботу стиснення пари при переведенні її від рівноважного тиску рв об'ємній фазі далеко від поверхні в область поверхневого шару з тиском насиченої пари р 0тоді вираз визначення потенціалу матиме вид:
ε = RT ln р 0 / р.
За допомогою такого рівняння можна перейти від координат x, p до координат ε і vта отримати криву, яка отримала назву «характеристична». Поляні у своїх дослідах виявив, що такі криві, побудовані за експериментальними даними отриманих ізотерм, мають таку властивість: вони інваріантні по відношенню до Т, або, інакше кажучи, всі криві такого типу можуть лягати на одну криву ε −ε .
Таке становище М. Поляні прийняв як постулат, тобто:
Зазначена властивість Поляні має величезне практичне значення, вона може по одній експериментальній ізотермі адсорбції побудувати сімейство ізотерм.
Теорія Поляні не дає аналітичного виразу для ізотерми або функції потенціалу від об'єму, але дозволяє обчислити координату будь-якої заданої температури, якщо відома хоча б одна ізотерма. Такий результат дуже важливий для технологічних розрахунків, тому що для подібних газів на одному адсорбенті криві адсорбції можуть бути близькими один до одного і можуть бути у багатьох випадках суміщені.
39. Характеристична крива адсорбції. Температурна інваріантність та афінність характеристичних кривих
Силове поле, яке виникає біля поверхні адсорбенту, багато в чому може бути схоже з гравітаційним полем. В адсорбційному полі можна уявити потенційні поверхні, тобто поверхні для яких характерний один і той же адсорбційний потенціал. Під поняттям адсорбційного потенціалу θ слід розуміти не що інше, як роботу, що виконується проти сил адсорбції при переміщенні 1 молячи адсорбтиву з певної точки поля в деяку газову фазу. Максимальний адсорбційний потенціал існуватиме на межі «адсорбент – адсорбційний обсяг». Але на межі «обсяг – газова фаза» (саме там закінчується дія адсорбційних сил) потенціал адсорбції повинен дорівнювати нульовому значенню. Зміна адсорбційного потенціалу за зміни адсорбційного обсягу можна як кривих. Вперше це зробив М. Поляні. Подібні типи кривих не залежать від температури і можуть бути характерними для кожного конкретного адсорбенту, такі типи кривих прийнято називати характеристичними кривими адсорбції. Теорія полімолекулярної адсорбції приймає, що для обсягу адсорбції застосовується рівняння стану газу. Отже, ізотерми, які характеризують залежність густини адсорбтиву від об'єму для різної температури, нагадують ізотерми залежності тиску від об'єму. При низькій температурі сили адсорбції на поверхні можуть викликати конденсацію пари в рідину певної густини. При нижчих температурах, ніж критична, при конденсації весь адсорбційний обсяг буде заповнений рідиною. У цьому випадку крива адсорбції йтиме майже паралельно осі абсцис, яка пов'язана з малою стисливістю рідини. Потім крива адсорбції на межі «об'єм – газова фаза» різко опускається вниз, і, відповідно, щільність адсорбтиву досягає значення деякої густини газової фази. При температурах вищих, ніж критична, адсорбтив може поводитися як ідеальний газ, і графік виражатиметься як ізотерма залежності для ідеального газу за умови, що pV = RT. За таких умов адсорбований газ матиме максимальну щільність біля самої поверхні адсорбенту і мати мінімальну при безпосередній близькості від газової фази. Причому в цьому випадку важливо відзначити, що густина адсорбтиву в адсорбційному шарі ніде не досягає густини самої рідини. І якщо температура дуже близька до критичної, залежність щільності від обсягу виражатиметься кривою, близькою на вигляд до ізотерми, яка описується рівнянням Ван-дер-Ваальса.При такому розкладі частина адсорбованої речовини перебуватиме в адсорбованому обсязі в рідкому стані, а частина адсорбованої речовини – у газоподібному. Тоді крива найбільш різко знижуватиметься в ділянці, яка відповідає переходу від рідини до газу. Якщо побудувати характеристичну криву за дослідною ізотермою адсорбції одного з адсорбтивів, а знаючи відповідні коефіцієнти афінності для якогось іншого адсорбтиву, можна знайти ізотерму адсорбції та побудувати її для іншого адсорбтиву. Потенційна теорія адсорбції дає можливість обчислити різні ізотерми адсорбції різних парів на тому самому адсорбенті, причому по характеристичній кривій, яка отримана з ізотерми адсорбції однієї пари, тому що співвідношення адсорбційного потенціалу не залежить від адсорбційних обсягів.
Афінність(від лат. affinis - "споріднений") - хроматографія за спорідненістю. Метод очищення та поділу білків заснований на їхній виборчій взаємодії з лігандом, ковалентно пов'язаним з інертним носієм (афінна хроматографія). Вимірювання афінності токсиканту до рецептора, по суті, є експериментальним вивченням залежності між кількістю речовини, що додається в інкубаційне середовище, і кількістю утворюється в результаті взаємодії токсикант-рецепторного комплексу.
Адсорбція(від лат. ad - на, при і sorbeo - поглинаю), зміна (зазвичай - підвищення) концентрації речовини поблизу поверхні поділу фаз ("поглинання на поверхні"). Причина адсорбції- ненасиченістю міжмолекулярних зв'язків поблизу поверхні, тобто. існуванням адсорбційного силового поля. Тіло, яке створює таке поле, називають адсорбентом, речовина, молекули якої можуть адсорбуватися, - адсорбтивом, вже адсорбована речовина - адсорбатом. Процес, зворотний адсорбціїназивається десорбцією.
Природа адсорбційного поля є різною. Якщо адсорбція зв'язна з ван-дер-ваальсовими зв'язками, то адсорбціюназивають фізичною. Якщо це валентні зв'язки, тобто. адсорбціяпроходить з утворенням поверхневих хімічних сполук, адсорбціюназивають хімічною, або хемосорбцією. Важливими рисами хемосорбціївиявляє: незворотність, високі теплові ефекти (сотні кДж/моль), активований характер. Існує безліч проміжних видів адсобрціїміж фізичною та хімічною адсорбцією. Наприклад, адсорбція, спричинена утворенням водневих зв'язків Також можливі різні види фізичної адсорбції. Найчастіше зустрічається виникнення дисперсійних міжмолекулярних сил тяжіння, у зв'язку з тим, що вони є приблизно постійними для адсорбентів з поверхнею будь-якої хімічної природи (неспецифічна адсорбція). Фізична адсорбціяможе бути викликана електростатичними силами (взаємодія між іонами, диполями або квадруполями); при цьому адсорбціявизначається хімічною природою молекул адсорбтиву (так звана специфічна адсорбція). Важливу роль також грає геометрія поверхні розділу. якщо поверхня є плоскою, то це адсорбціявідкритої поверхні, у разі слабко або сильно викривленої поверхні - про адсорбціїу порах адсорбенту.
В теорії адсорбціїрозрізняють статику (система адсорбент-адсорбат знаходиться у термодинамічній рівновазі) та кінетику (рівноваги немає).
Статика адсорбції
Термодинаміка адсорбції
.Основи термодинаміки адсорбціїбули створені Дж.Гіббсом у 70-ті роки. ХІХ ст. За Гіббсом, у рівноважній двофазній системі поблизу поверхні розділу фаз відбувається деяка зміна локальних значень всіх екстенсивних властивостей (крім об'єму). Однак фази вважаються однорідними аж до деякої геометричної поверхні, що їх розділяє. Тому значення будь-якої екстенсивної властивості для системи в цілому не дорівнює сумі значень цієї властивості в однорідних фазах і . Різниця приписується двовимірної поверхневої фази, пов'язаної з поверхнею, що розділяє. Т.к. поверхнева фаза не має товщини, то V 0=+ та =-, де V-Об `єм.
Викладені уявлення дозволяють навести фундаментальне термодинамічний рівняння до виду:
де G-гіббсова вільна енергія, S-ентропія, - міжфазний поверхневий натяг, s-площа поверхні розділу, і n i- відповідний хімічний потенціал та кількість молей i-Того компонента. Індекс вказує на значення відповідної властивості поверхневої фази. Перетворення Лежандра дозволяє змінити рівняння (1) для ізотермічних умов:
Величина називається гіббсовою адсорбцієюі позначається символом Г (виражається в моль/см 2). Для двокомпонентної системи:
Положення розділяючої поверхні може бути вибрано довільно. Зокрема, вибір цього становища може задовольняти умові Р 1 =0. Така поверхня називається еквімолекулярною. Для неї вводиться позначення Г2 = Г2(1). Звідси випливає основне адсорбційне рівняння Гіббса:
Якщо адсорбтив зовсім не розчинний в одній із двох фаз, =const, і перехід від рівняння (2) до рівняння (3) не вимагає умови Г 1 =0. Таким чином, гіббсова адсорбція- це надлишок даного компонента в реальній двофазній системі в порівнянні з такою системою, в якій обидві фази були б однорідні аж до розділяючої поверхні. Крім гіббсових надлишкових величин адсорбції, в її теорії велику роль відіграє адсорбція, що розуміється як повний зміст компонента iв просторі W, В якому виявляються адсорбційні сили. Позначаючи повний зміст через аі вважаючи, що компонент iзовсім не розчинний в одній з об'ємних фаз, маємо:
де c i-концентрація i-Того компонента в об'ємній фазі. При малих з i:
Адсорбціяможе відбуватися на будь-якій поверхні розділу між двома будь-якими фазами, зокрема на поверхні розділу флюїд-флюїд (рідина-газ, рідина-рідина) або тверде тіло-флюїд (тверде-газ, тверде-рідина). У системах флюїд-флюїд можна виміряти як функцію і експериментально визначити Г 2 (1) за рівнянням (3). У другому випадку для визначення Г 2 (1) вимірюють будь-яким методом n i 0 , і концентрації i-того компонента в цих обсягах. Звідси обчислюють Г i(1). Цей метод називається об'ємним (або волюмометричним). При ваговому (гравіметричному) методі безпосередньо визначають кількість i-Того компонента на поверхні розділу.
Ізотерма адсорбції
.У рівноважній адсорбційній системі параметри, що визначають рівновагу, це a iпарціальний тиск р(або з i) і температура Т. Вони пов'язані так званим термічним рівнянням:
При адсорбціїіндивідуального адсорбтиву ( i=1) це рівняння набуває вигляду:
Три окремі випадки термічного рівняння (коли Т, рабо a- Константи) відіграють особливу роль у теорії адсорбції:
а=- рівняння ізотерми адсорбції,
Т=- Рівняння ізабори адсорбції,
Р-- рівняння ізостери адсорбції.
Конкретний вид функцій визначається особливостями аналізованої системи. Якщо одна з них, наприклад, відома для будь-якого значення Т= const, то, очевидно, стають відомими і дві інші. При цьому не обов'язково, щоб був відомий аналітичний вид залежностей. Вони можуть бути задані емпірично у вигляді набору значень а, рі Т.
В теорії адсорбціїзазвичай вирішується питання про вид функції а=(р) р, тобто. про рівняння ізотерми адсорбції. Ця проблема пов'язана з тепловими ефектами, що супроводжують адсорбцію. При розрахунку зміни значень основних термодинамічних функцій у разі переходу dnмолей адсорбтиву з об'ємної фази в поверхневу в рівноважній системі при р = const можливі два випадки: у першому враховується тільки перетворення адсорбтиву на адсорбат, оскільки адсорбент при адсорбціїтермодинамічно незмінний та її роль - служити джерелом адсорбційного поля; у другому враховується та зміна адсорбенту.
Оскільки система рівноважна, то хімічні потенціали адсорбату та адсорбтиву однакові; ентропія адсорбату внаслідок зменшення рухливості молекул при адсорбціїменше ентропії адсорбтиву. Тому за інертному адсорбенті ентальпія завжди негативна, тобто. адсорбціяекзотермічна. Врахування зміни ентропії адсорбенту може змінити цей висновок. Наприклад, при сорбції полімерами речовин, в яких набухає полімер, ентропія останнього (через збільшення рухливості макромолекул) може настільки сильно зрости, що адсорбціястає ендотермічною. Надалі у статті розглядається лише екзотермічна адсорбція.
Розрізняють інтегральну, диференціальну, ізостеричну та середню теплоти адсорбції. Інтегральна теплота Qдорівнює спаду ентальпії (при V= const - постійної внутрішньої енергії) при зміні адсорбціївід a 1до а 2(В окремому випадку може бути а 1 = 0): Q= -(Н 2 - Н 1). Цю величину зазвичай відносять до маси адсорбенту і виражають у Дж/кг.
Диференційна теплота q(Дж/моль) дорівнює спаду ентальпії dHпри зміні ана da. Її висловлюють ставленням q = - (dH/da). Очевидно, що
Ізостеричну теплоту q st приймають рівною:
де - різниця мольних обсягів адсорбату та адсорбтиву. Можна показати, що  для ідеального газового адсоритиву:
для ідеального газового адсоритиву:
Сенс запровадження q siу тому, що для її визначення не потрібні калориметричні дані (такі, як Qі q) і вона може бути обчислена за рівнянням (9) за результатами виміру адсорбції. Вводять також середню теплоту Q(Дж/моль):
Зі зростанням апараметр Qзавжди зростає, a qможе зменшуватися, збільшуватися чи залишатися незмінною. Зі зростанням апри неоднорідній поверхні адсорбціявідбувається на менш активних ділянках, що призводить до зменшення q. Однак при цьому зменшуються середні відстані між адсорбованими молекулами, внаслідок чого збільшуються сили тяжіння між ними, та qзростає. Співвідношення між двома згаданими ефектами визначає перебіг залежності q=f(a). При дуже великих апочинають переважати сили відштовхування і у цій галузі qзавжди знижується зі зростанням a.
При дуже малих заповненнях поверхні рівняння ізотерми адсорбціямає вигляд рівняння Генрі:
де К H – коефіцієнт Генрі. Дійсно, за дуже малих аадсорбційний шар подібний до двомірного ідеального газу, тому його рівняння стану має вигляд: = RT,де - двомірний тиск, - площа, яку займає одним молем речовини. Звідси, враховуючи, що =- і використовуючи рівняння (3), отримуємо рівняння (12). Рівні Генрі вимагає, щоб qбуло постійним. При великих заповненнях це рівняння перестає виконуватись. Тому Г.Фрейндліх (1906) запропонував описувати ізотерми. адсорбціїнаступним емпіричним рівнянням (рівняння Фрейндліха):
де kі n- Константи. Цим рівнянням часто користуються як інтерполяційною формулою, хоча воно за малих рне переходить у рівняння (12), а за дуже великих рпризводить до неузгодженого з досвідом необмеженого зростання а.
Сувора теорія ізотерми адсорбціябула створена І. Ленгмюром (1914-18). В основу теорії покладено слід. модель: 1) поверхня адсорбенту є набір енергетично однакових активних центрів, на яких адсорбуються (локалізуються) молекули адсорбтиву; 2) одному центрі адсорбується лише одна молекула, тобто. при адсорбціяутворюється лише один адсорбц. шар (моношар); 3) адсорбціяна даному центрі не впливає адсорбціяна ін. центрах, тобто взаємод. адсорбованих молекул можна знехтувати.
Модель Ленгмюра зв. локалізованої мономолекулярної адсорбціяна однорідній поверхні. рівняння ізотерми адсорбція, що відповідає цій моделі, м.б. отримано за допомогою разл. методів (молекулярно-кінетич., Термодинамічні., Статистико-термодинаміч.). Так, адсорбції. рівновагу можна висловити слід. схемою:
Молекула Віль. Адсорбці. у газовій + адсорбц. комплекс фазі центр (зайнятий центр)
Концентрація молекул у газі пропорційна р, концентрація вільн. центрів-величині ( а т - а),де а т -повне число центрів, а число зайнятих центрів, концентрація адсорбц. комплексів-величині адсорбціяОтже, константа рівноваги дорівнює: К р = р (а т -а)/ адсорбціяЗвідси отримуємо рівняння Ленгмюра:
де b-Т. зв. адсорбції. коеф., рівний К р -1.В області дуже малих тисків bр " 1 та a = (a m b)p,що відповідає рівнянню Генрі, в якому До H= a m b.В області дуже великих тисків bр 1 та аа т;при цьому адсорбціяперестає залежати від тиску. Константа рівноваги b -1пов'язана зі стандартним значенням ізобарного потенціалу реакції:
Модель Ленгмюра вимагає диференціації. теплота та ентропія адсорбціяне залежали від ступеня наповнення поверхні.
рівняння (14)-суворий вираз, що відповідає моделі Ленгмюра, проте воно рідко виправдовується на досвіді, оскільки сама модель ідеалізована адсорбціяВчення про адсорбціяз 20-х років. 20 ст. отже. ступеня будувалося на основі ослаблення або виключення того чи іншого припущення Ленгмюр адсорбція
Вже Ленгмюр запропонував спосіб опису адсорбціяна неоднорідній поверхні (тобто при припущенні, що не всі центри однакові). Об'єднуючи однакові центри в групи і вважаючи, що до кожної групи застосовується рівняння (14), можна вважати, що адсорбціяна всій поверхні виражається сумою членів рівняння (14):
Вважаючи, що число адсорбції. центрів М.Б. описано безперервною ф-цією розподілу за значеннями вільн. енергії, Я.Б.Зельдович отримав з ф-ли (16) для експоненційної ф-ції рівняння типу (13).
адсорбціяна неоднорідних поверхнях - великий розділ теорії адсорбціяЇї осн. задача-рішення інтегрального рівняння:
де f(р)- т. зв. емпірич. ізотерма адсорбція, -Та чи інша ф-ція розподілу числа центрів за значеннями своб. енергії, ( b, р)-локальна ізотерма адсорбція, в кач-ве якої зазвичай приймають ізотерму Ленгмюр адсорбція
Багато спроб зроблено у напрямку відмови від другого припущення Ленгмюр адсорбціяНа цьому шляху особливого значення набула теорія полімолекулярної адсорбція, запропонована С. Брунауером, П. Емметом та Е. Теллером (теорія БЕТ) Теорія постулює, що при температурі нижче критичної кожна молекула, адсорбована в першому шарі (теплота адсорбції) q i,) є центром для молекул, що утворюють другий шар, і т.д. При цьому вважається, що теплота адсорбціяу всіх шарах, крім першого, дорівнює теплоті конденсації Така модель призводить до рівняння:
де с =ехр [(q 1 -) / RT]. рівняння (18) у координатах a, p/p sвідповідає S-подібної кривої. у координатах p/p s ,
ізотерма адсорбціяза рівнянням (18) має бути лінійною. Нахил цієї прямої (зазвичай в інтервалі 0,05 p/p s 0,30) і відрізок, що відсікає нею на осі ординат, дають значення соотв. а ті с.Широке поширення теорії БЕТ пов'язано з тим, що її автори фактично вважають адсорбціянелокалізованою, ототожнюють константу а тне з числом дискретних адсорбцій. центрів, а з числом молекул адсорбату в першому шарі при щільній упаковці (при р= p s).Тому, вводячи уявлення про площу, яку займає одна молекула в цьому шарі, приймають:
де s-площа поверхні адсорбат адсорбціяЯк правило, для цього вимірюють ізотерму адсорбціяазоту і приймають, що з його молекулы= 0,162нм 2 . Часто виконуваний аналогічний розрахунок sза моделлю Ленгмюра не коректний, т.к. цей метод, очевидно, застосовується тільки до нелокалізованої адсорбція
У теорію полімолекулярної адсорбціявеликий внесок вніс Я. де Бур, експериментально показав, що залежність середньої кількості шарів (понад перший) всіх поверхнях, близьких по хімічним. природі, від p/p sвиражається універсальною кривою (т. зв. t-кривою). Це також дозволяє оцінювати площу поверхні адсорбтивів.
Робилися спроби врахувати у моделі Ленгмюра також взаємодій. між адсорбіром. молекулами. Так, Т. Хілл та Я. де Бур, вважаючи, що рівняння стану адсорбц. шару є двомірний аналог рівняння Ван-дер-Ваальса, що отримали слід. рівняння ізотерми адсорбція:
де = а/а т, аі b-константи рівняння Ван-дер-Ваальс адсорбціяР. Фаулер та Е. Гуггенгейм, врахувавши взаємод. адсорбірів. молекул, вивели рівняння:
де-константа, пов'язана з парною взаємодією молекул.
Існує ще один механізм, що призводить до доповнення. адсорбціяадсорбтивів нижче їх критич. температури на пористих адсорбентах при порівняно високих значеннях p/p s.Це – капілярна конденсація. Якщо в порі утворився увігнутий меніск адсорбату, то в ній починається конденсація при p/p s Відповідно до рівняння Кельвіна:
де-поверхневий натяг адсорбату, V -йогомольний об'єм, r-радіус кривизни меніск адсорбціяКапілярна конденсація призводить до різкого підйому ізотерми. адсорбціяУ цьому часто (але завжди) спостерігається т. зв. адсорбції. гістерезис, тобто. розбіжність адсорбц. та десорбц. гілок ізотерми. Як правило, це пов'язано з тим, що форми менісків при адсорбціята десорбції не збігаються.
Капілярну конденсацію використовують для визначення розмірів пор адсорбент адсорбціяПо рівнянню (22) для кожного значення p/p sобчислюють радіус кривизни меніск адсорбціяЗ нього, враховуючи товщину адсорбції. шару (напр., по t-кривий), форму перехідної області від шару до меніска та залежність від кривизни при дуже малих r , знаходять лінійний розмір (ефективний радіус r ef) пір, що заповнюються при цьому p/p s.Обсяг таких пір визначають за приростом адсорбціяу цій точці ізотерми. Використовуючи отримані дані, будують криву розподілу обсягу пор за їхніми радіусами. Метод застосовується при r ef 1,5 нм. Зазвичай розрахунок ведуть по десорбції. гілки ізотерми, але суворіша совр. Теорія вимагає для побудови кривої обліку обох гілок.
Потенційна теорія адсорбції та теорія об'ємного заповнення мікропор.Модель адсорбція, принципово відмінну від ленгмюрівської, запропонував у 1914 р. М. Поляки. Відповідно до цієї моделі, поблизу поверхні адсорбенту існує потенційне адсорбц. силове поле, що зменшується з відстанню від поверхні. Внаслідок цього тиск адсорбтиву, рівний далеко від поверхні р, поблизу неї зростає і на деякій відстані досягає значення p s, при якому адсорбтив конденсується. Об'єм шару між поверхнею розділу і геом. місцем точок, де р = p s заповнений рідиною, якій приписуються нормальні значення фіз. властивостей об'ємної рідини Оборотна ізотерміч. робота е адсорбц. сил, що визначається за рівнянням = RTlnp/ps, зв. адсорбції. потенціалом, а вся концепція-потенційною теорією адсорбціяПри заданій величині обсягу Vадсорбції. шару потенційно залежить від температури (внаслідок незалежності дисперсійних сил від температури). Така температурна інваріантність дає можливість перераховувати адсорбціяз однієї т-ри на іншу, хоча рівняння ізотерми адсорбціяна основі теорії, що викладається, вивести не вдавалося. Модель Поляні широко та успішно застосовувалася багато. авторами, однак вона містила два дуже вразливі положення: 1) припущення про те, що найтонша адсорбція. плівка має нормальні значення фіз. властивостей об'ємної рідини (це припущення не підтверджувалося дослідами); 2) температурна інваріантність ф-ції =f(V),лежача в основі теорії, приблизно підтверджувалася досвідом тільки для дуже тонкопористих адсорбентів.
Використовуючи потенційну теорію, М.М. Дубінін запропонував та розробив теорію об'ємного заповнення мікропор (ТОЗМ). Було постулировано, що це теорія застосовна лише мікропористим адсорбентам. Особливість таких адсорбентів, у яких лінійні розміри пір r1 нм, полягає в тому, що весь обсяг їх пір "заповнений" адсорбц. полем. Тому при адсорбціявони заповнюються не пошарово, а об'ємно. Величина в даному випадку - це не адсорбц. потенціал, а з точністю до хімічного знака. потенціал адсорбату, що відраховується від рівня хім. потенціалу нормальної рідини за тієї ж температури. Вся сукупність пор адсорбентів поділяється на три класи: мікропори ( r 0,6 нм), мезопори (0,6 нмr20 нм) та макропори ( r 20 нм). адсорбціяу мікропорах відбувається за схемою ТОЗМ, тобто. об'ємно, в мезопорах-за механізмом пошарового заповнення, що завершується капілярною конденсацією. Макропори при адсорбції. рівноваги жодної ролі не грають.
Ввівши уявлення про ф-ції розподілу обсягів пор за значеннями хім. потенціалу адсорбату в них, М.М. Дубінін та Л. В. Радушкевич отримали рівняння ізотерми адсорбції ТОЗМ, яке зазвичай записують у слід. формі:

де п, Еі а 0 -параметри ( а 0 = апри р = p s). Температурна залежність a 0:
де = -(Da 0 /dT); a 0 0 = a 0при Т = Т0. Параметри пі Епрактично не залежить від температури. В більшості випадків п= 2. Тільки випадків, коли початкові теплоти адсорбціядуже великі, п>2.Для перерахунку ізотерм адсорбціяз одного адсорбтиву на інший приблизно допускають, що E 1 /E 2 P 1 /P=і що a 01 /a 02 V 1 /V 2 де P i-парахор, V i -мольний обсяг адсорбтив адсорбція
Кожен мікропористий адсорбент характеризується за ТОЗМ двома параметрами: W-об'ємом мікропор ( W 0 = = a 0 V 0)і E 0 -характеристич. енергією; W 0та E 0 відносять до стандартного адсорбтиву, зазвичай до бензолу.
Користуючись уявленням, що у реальному адсорбенті є пори різних розмірів, і запроваджуючи розподіл значень Е здисперсією, що дорівнює Ф. Стеклі запропонував узагальнення рівняння (23), назване рівнянням Дубініна-Стеклі:

де B 0 -константа, пов'язана з Eу рівнянні (23), а у= ![]() . T.к. в адсорбції. технікою наиб. поширення набули саме мікропористі адсорбенти (активне вугілля, цеоліти, тонкопористі ксерогелі), ТОЗМ застосовується не тільки у фіз.-хім. дослідженнях, а й у інженерних розрахунках.
. T.к. в адсорбції. технікою наиб. поширення набули саме мікропористі адсорбенти (активне вугілля, цеоліти, тонкопористі ксерогелі), ТОЗМ застосовується не тільки у фіз.-хім. дослідженнях, а й у інженерних розрахунках.
Адсорбція газових та рідких сумішей. На практиці завжди мають справу не з індивідуальним адсорбтивом, а із сумішшю газів або з рідкими розчинами. Тому потрібне узагальнення теорії адсорбціяна випадок багатокомпонентного адсорбтиву адсорбціяВ принципі можна виходити з будь-якої моделі адсорбціята поширити її на цей випадок. При адсорбціягазової суміші це досягається не лише великим ускладненням рівнянь, а й уведенням у них доповнить. емпірич. параметрів, пов'язаних або з взаємод. різнорідних молекул або, у загальному вигляді, із впливом одних в-в на коэф. активності інших. Тільки модель Ленгмюра дозволяє отримати рівняння ізотерми адсорбціясуміші без параметрів, що не входять до рівнянь для адсорбціяіндивідуальних ст-в. Для цього достатньо врахувати, що при адсорбції k-того компонента із суміші iкомпонентів частина адсорбції. центрів М.Б. зайнята ін молекулами. Тому:
В разі адсорбціярідких розчинів незалежно від їх концентрації вся поверхня адсорбенту заповнена адсорбціяВнаслідок цього адсорбціямолекули k-того компонента супроводжується витісненням деякого числа молекул інших компонентів, тобто. адсорбціяносить конкурентний характер.
Розрізняють молекулярну та іонну адсорбціярозчинів. Перша відбувається за адсорбціярозчинів неелектролітів, друга-р-рів електролітів. Молекулярна адсорбція, як правило, виражається надмірними величинами. Конкурентний характер адсорбціяобумовлює те, що величина ам.б. як позитивною, і негативною. Висловлюючи адсорбція i- того компонента як ф-цію його мольної частки в розчині х i-, маємо, що Г i= Про при х i= 0 і х i= 1 (можливою зміною обсягу речовини в адсорбц. шарі нехтують). Тому ізотерма адсорбціямає один або дек. екстремумів.
рівняння ізотерми адсорбціябінарних розчинів неелектролітів, надійно обґрунтованих термодинамічно, має вигляд:

де індекс s вказує на адсорбції. фазу, - ( dn s 2 /dn s 1)показує, скільки молей другого компонента витісняється одним молем першого,-різниця доданків (стандартних частин) хімічних. потенціалу, що залежить тільки від температури.
Осн. проблема використання цього та ряду ін. рівнянь ізотерми адсорбція-З'ясування залежності коеф. активності компонентів в адсорбції. шарі від його складу адсорбціяНайважливіше питання при застосуванні адсорбціядля поділу чи очищення речовин-підбір селективного адсорбенту по відношенню до даного компоненту розчин адсорбція
Іонна адсорбція, як правило, не носить еквівалентного характеру адсорбціяНа поверхні з розчину електроліту адсорбуються переважно. катіони або аніони. Завдяки електрич. (кулонівським) силам на поверхні утворюється подвійний електричний шар.
Якщо до складу адсорбенту входять іони або поверхневі функції. групи, здатні в даному розчиннику до іонізації, між адсорбентом і розчином електроліту відбувається іонний обмін. Адсорбент у разі наз. іонітом.
Кінетика адсорбції
адсорбція, Як і будь-який реальний процес, відбувається у часі. Тому повна теорія адсорбціяповинна містити розділ про кінетику адсорбціяЕлементарний акт адсорбціяздійснюється практично миттєво (виняток-хемосорбція). Тому тимчасові залежності адсорбціявизначаються в осн. механізмом дифузії, тобто підведення адсорбтиву до місця адсорбціяЯкщо адсорбціяна відкритій поверхні не миттєва, такий процес відбувається у зовнішньодифузійній ділянці; при цьому закони дифузії не специфічні для адсорбціяУ разі пористих адсорбентів, крім внеш. дифузії, важливу роль починає грати всередину. дифузія, тобто. перенесення адсорбтиву в порах адсорбенту за наявності у яких градієнта концентрації. Механізм такого перенесення може залежати від концентрації адсорбтиву та розмірів пір.
Розрізняють молекулярну, кнудсенівську та поверхневу (фольмерівську) дифузію. Молекулярна дифузія здійснюється, якщо довжина вільн. пробігу молекул у порах менше розміру пір, кнудсенівська-якщо ця довжина перевищує розмір пір. При поверхневій дифузії молекули переміщуються поверхнею адсорбенту без переходу в об'ємну фазу. Проте значення коеф. дифузії не однакові для різних механізмів дифузії. У багатьох. випадках експериментально не вдається встановити, як відбувається дифузія, і тому вводять т. зв. ефективний коеф. дифузії, що описує процес загалом.
Осн. Експерім. матеріалом про кінетику адсорбціяслужить т. зв. кінетич. крива, тобто. ф-ція = а/а дорівнює = f(t) де-відносна адсорбція, що дорівнює відношенню поточного значення адсорбції адо aрівн - її значення при часі t.Для тлумачення кінетич. кривою у найпростішому разі припускають, що зерно адсорбенту має абсолютно однорідну за обсягом пористу структуру (цю модель зв. квазігомогенній). значить. удосконалення квазигомогенної моделі-уявлення про те, що кожне зерно містить області з більшими та більш тонкими порами. Дифузія у такому зерні описується двома разл. коефіцієнтами.
У разі відкритої поверхні, приймаючи модель Ленгмюра, легко отримати кінетич. рівняння адсорбціяШвидкість наближення до рівноваги є різницею швидкостей адсорбціята десорбції. Вважаючи, як завжди в кінетиці, що швидкості процесів пропорційні концентраціям реагують в-в, маємо:
де k адс і k дес - константи швидкості соотв. адсорбціята десорбції. Тиск у газовій фазі вважається незмінним. При інтегруванні цього рівняння від t= 0 до будь-якого значення tотримаємо:
Звідси при f маємо: = рівн. Тож остаточно маємо:
де k = k адс + k дес.
Вплив температури на швидкість адсорбціявиражається рівнянням, аналогічним рівнянню Арреніус адсорбціяЗі збільшенням температури k адс експоненційно зростає. Т.к. дифузія в порах адсорбенту пов'язана з подолання активації. бар'єрів, температурні залежності k адс і k дес не однакові.
Знання швидкостей дифузії важливо як для теорії адсорбція, але й розрахунку пром. адсорбції. процесів. При цьому зазвичай мають справу не з окремими зернами адсорбенту, а з їх шарами. Кінетика процесу у шарі виражається дуже складними залежностями. У кожній точці шару на даний момент часу величина адсорбціявизначається не лише видом рівняння ізотерми адсорбціяі закономірностями кінетики процесу, але також аеро- або гідродинаміч. умовами обтікання зерен газовим чи рідинним потоком Кінетика процесу у шарі адсорбенту на відміну кінетики в окремому зерні зв. динамікою адсорбція, загальна схема розв'язання задач якої така: складається система диференц. рівнянь у приватних похідних, що враховує характеристики шару, ізотерму адсорбція, дифузійні характеристики (коеф. дифузії, види перенесення маси по шару і всередині зерен), аеро- та гідродинаміч. особливості потік адсорбціяЗадаються початкові та крайові умови. Вирішення цієї системи рівнянь у принципі призводить до значень величин адсорбціяна даний момент часу в цій точці шару. Як правило, аналітич. рішення вдається отримати лише найпростіших випадків, тому таке завдання вирішується чисельно з допомогою ЕОМ.
При дослідному вивченні динаміки адсорбціячерез шар адсорбенту пропускають газовий або рідинний потік із заданими характеристиками та досліджують склад вихідного потоку як ф-цію часу. Поява поглинається за шаром зв. проскоком, а час до проскоку – часом захисної дії. Залежність концентрації даного компонента за шаром від часу зв. вихідний кривій. Ці криві служать осн. Експерім. матеріалом, що дозволяє судити про закономірності динаміки адсорбція
Апаратурне оформлення адсорбційних процесів
Існує безліч технол. прийомів проведення адсорбції. процесів. Широко поширені цикліч. (періо-дич.) установки з нерухомим шаром адсорбенту, осн. вузол яких - один або дек. адсорберів, виконаних у вигляді порожніх колон, що заповнюються гранульованим адсорбентом. Газовий (або рідинний) потік, що містить компоненти, що адсорбуються, пропускається через шар адсорбенту до проскок адсорбціяПісля цього адсорбент в адсорбер регенерують, а газовий потік направляють в ін. адсорбер. Регенерація адсорбенту включає ряд стадій, у тому числі основна-десорбція, тобто. виділення раніше поглиненого в-ва з адсорбенту адсорбціяДесорбцію проводять нагріванням, скиданням тиску в газовій фазі, витісненням (напр., гострою водяною парою) або комбінацією цих методів. Т. до. часи адсорбціяі регенерації не збігаються, підбирають таке число одночасно працюючих і адсорберів, що регенеруються, щоб в цілому процес йшов безперервно.
За техн. та економіч. міркувань регенерацію не доводять до кінця адсорбціяТому робоча ємність адсорбенту дорівнює різниці між максимально досягається в даних умовах адсорбціяі кол-вом адсорбату, що залишається в адсорбенті після регенерації. Внаслідок цього ізотерми адсорбція, що відповідають процесу в адсорбері, не повинні бути надто крутими.
В описаній схемі можливі два варіанти: 1) цільовий продукт адсорбується з газового потоку практично повністю, і тоді він міститься в десорбаті, звідки його витягують тим чи іншим способом; 2) цільовий продукт адсорбується гірше, ніж ін. компоненти газової суміші, і тоді він міститься у газовому потоці, що виходить. За першим варіантом працюють, наприклад, рекупераційні установки на віскозних виробах, що вловлюють з відхідних газів і повертають в цикл CS 2 . Продуктивність таких установок досягає сотень тисяч м 3 газу, що очищається на годину; адсорбент-активне вугілля з дуже тонкими микропорами, тобто. вугілля, в якому константа EТОЗМ (див. вище) дорівнює 20-25 кДж/моль. Це значення E 0 відповідає не надто крутій ізотермі, що забезпечує добрі умови регенерації. Таке вугілля зв. рекупераційними. Десорбцію здійснюють гострим водяною парою. Для економії енергії холодні та газові гарячі потоки пропускають через теплообмінники.
Дуже важлива осушка газів і рідин, наприклад нафтових газів перед їх переробкою або прир. газів перед транспортуванням; адсорбенти-силікагель чи цеоліти. Десорбцію здійснюють нагріванням. Т. до. десорбція цеоліту пов'язана з великими енерговитратами, застосовують комбінований адсорбент: осн. масу вологи поглинають легко регенерується силікагелем, а глибоку доосушування-цеоліт.
При тепловій регенерації повний цикл включає адсорбція, нагрівання адсорбенту, його десорбцію та охолодження Велика кількість стадій обумовлює низьку інтенсивність і високу енергоємність адсорбціяТому стала вельми поширеною отримали т. зв. короткоциклові установки, весь цикл у яких займає дек. хвилин. Вони газ подається в адсорбер під значить. тиском, який потім скидається, і відбувається десорбція. Весь процес іде майже ізотермічно (відхилення від ізотермічності викликається лише виділенням теплоти адсорбціята поглинанням теплоти при десорбції). Стадії циклу: адсорбція, скидання тиску, десорбція, підйом тиску. Приклад установки з цеолітом для отримання повітря, збагаченого киснем.
В установках шаром адсорбенту, що зсувається (в т. зв. гіперсорберах) останній під дією сили тяжіння повільно опускається, виводиться з ниж. частини адсорбера і потрапляє у т. зв. ерліфт, що являє собою вертикальну трубу, паралельну адсорбції. колон. По цій трубі знизу вгору рухається потік повітря, який піднімає збіжжя адсорбенту вгору. частина колони. Газовий потік, що переробляється, надходить у середню частину адсорбера і рухається вгору протитечією до адсорбенту. У верхній частині колони безперервно відбувається адсорбція, у нижній - регенерація адсорбенту (див. також Адсорбційне очищення).
В установках з псевдозрідженим ("киплячим") шаром адсорбенту газовий потік, що надходить до адсорберу знизу, приводить адсорбент у зважений стан. При цьому різко збільшується ефективність масообміну між адсорбентом та газом та скорочується тривалість адсорбціята десорбції. Такі установки мають високу продуктивність. Їх широкому поширенню перешкоджають високі вимоги до хутра. міцності зерен адсорбенту (недостатня міцність обумовлює значить. втрати адсорбенту внаслідок його стирання та винесення з апарату).
Осн. вимоги до адсорбентів: велика адсорбц. ємність, тобто. вони повинні бути дисперсні тіла з великою уд. поверхнею або з великим обсягом пор; хім. природа поверхні має забезпечувати ефективну адсорбціяданих у даних умовах; хім. та терміч. стійкість, регенерованість, доступність. наиб. поширення набули активні вугілля, ксерогелі деяких оксидів (силіка-гелі, алюмогелі та ін.), цеоліти; із непористих адсорбентів-техн. вуглець (сажа) та високодисперсний SiO 2 (аеросил, "біла сажа").
Області застосування адсорбційної техніки
на явищі адсорбціязасновані багато. способи очищення повітря від шкідливих домішок (див. Газів очищення),води (див. Водопідготовка),а також цукрових сиропів при цукроваренні, фруктових соків та ін рідин в їжу. пром-сті, відпрацьованих мастил. Видалення вологи як шкідливої домішки з газів та рідин за допомогою твердих адсорбентів-одна з важливих галузей адсорбції. техніки (див. також Газів осушка).
На адсорбції. процесах засновано тонкий поділ сумішей речовин та виділення зі складних сумішей певних компонентів. Приклади-поділ ізомерів алканів з метою отримання нормальних вуглеводнів для виробництва ПАР, поділ нафт при виробництві моторних палив. Для газових сумішей адсорбції. методи поділу використовують при отриманні повітря, збагаченого киснем (аж до майже чистого 2); у мн. випадках ці методи успішно конкурують із ректифікаційним (див. Повітря розподіл).
Область застосування адсорбц, що швидко розвивається. техніки-медицина, де вона служить для вилучення шкідливих речовин із крові (метод гемосорбції) та ін. фізіол. рідин. Високі вимоги до стерильності ставлять дуже складне завдання підбору відповідних адсорбентів. До них відносяться спеціально підготовлені активні вугілля.
Літ.:Брунауер С., Адсорбція газів та пари, пров. з англ., Т. 1, М., 1948; де Бур Я, Динамічний характер адсорбції, пров. з англ., М., 1962; Адсорбція та пористість, під ред. М. М. Дубініна [та ін], М., 1976; Кельієв Н. Ст, Основи адсорбційної техніки, 2 видавництва, М., 1984; Young D.M., Crowell A.D., Physical adsorption of gases, L., 1962. М.М.Дубінін, В.В. Серпінський.
Виберіть першу літеру у назві статті:
У разі взаємодії двох атомів:
U – енергія взаємодії;
U = U ПРИТЯЖ. + U ВІДТАЛ.
 - рівняння Леннарда-Джонса
, c, b, m = const
- рівняння Леннарда-Джонса
, c, b, m = const
У випадках взаємодії атомів з твердою поверхнею необхідно провести підсумовування всіх взаємодій.
х– відстань до поверхні
r – радіус дії сил тяжіння
dV – обсяг
n – число молекул поверхні
U АДС. – енергія адсорбційної взаємодії



У разі адсорбції тяжіння посилюється. І у разі при взаємодії типу неполярно-неполярна адсорбція переважно локалізується у заглибленнях.
Електростатична взаємодія.
Полярний адсорбент – неполярний адсорбат
Неполярний адсорбент – полярний адсорбат
Полярний адсорбент – полярний адсорбат.
М  олекулу адсорбату представляють як диполь, а адсорбенту – як провідник, у якому молекула адсорбату індукує диполь дзеркально симетрично стосовно даного.
олекулу адсорбату представляють як диполь, а адсорбенту – як провідник, у якому молекула адсорбату індукує диполь дзеркально симетрично стосовно даного.
X – відстань до середини
При взаємодії виникає потенціал:
 ,
,
 - Дипольний момент.
- Дипольний момент.
Потенціал прагне набути максимального значення, тобто. диполі прагнуть зорієнтуватися перпендикулярно поверхні.
Оскільки підвищення температури сприяє зростанню броунівського руху, воно призводить до гальмування процесу адсорбції.
У разі електростатичної взаємодії адсорбат переважно локалізується на виступах.
Фундаментальне адсорбційне рівняння.
У разі адсорбції відбувається перерозподіл компонента, отже, змінюється хімічний потенціал. Процес адсорбції можна як перехід поверхневої енергії в хімічну.
Об'єм шару = 0, тоді узагальнене рівняння I та II закону термодинаміки:
T = const; (1) = (2) => 

Для двокомпонентної системи:
,
 ,
,
 =>
=>
=>
 - адсорбційне рівняння Гіббса
.
- адсорбційне рівняння Гіббса
.
Для випадку адсорбції тв. тіло – газ: , 
 ,
,

 - Ізотерма
- Ізотерма
 - ізобара
- ізобара
 - ізопікна
- ізопікна
 - ізостера
- ізостера
Ізотерма, ізопікна, ізостера пов'язані один з одним.
Т.к. адсорбція функція 



Ізотерма Генрі Ізотерма Лангмюра



Термодинаміка. Адсорбція.
Для конденсованих середовищ:
 ,
,
 ,
,
 - інтегральна зміна енергії Гіббса
.
- інтегральна зміна енергії Гіббса
.

P-тиск над викривленою поверхнею, P S-тиск над плоскою поверхнею
 - адсорбційний потенціал
- адсорбційний потенціал
Диференційна зміна ентрапії 
 , Г = const
, Г = const
- диференційна зміна ентропії
- диференціальна ентальпія адсорбції
 - ізостерична теплота адсорбції
- ізостерична теплота адсорбції
 - теплота конденсації
- теплота конденсації
 - чиста теплота адсорбції
- чиста теплота адсорбції

 ,
,




Qa - інтегральна теплота адсорбції, 
Qra – інтегральна чиста теплота адсорбції,

Рівняння Генрі
Дослідження адсорбції ускладнюється неоднорідністю поверхні, тому найпростіші закономірності набувають для однорідних поверхонь.
Розглянемо взаємодію газів з твердою поверхнею, коли здійснюється перехід газу з рівноважного стану в обсязі рівноважний стан на поверхні. Цей випадок аналогічний рівновазі газів у полі сили тяжіння.
 ,
,
 ,
=>
,
=> -рівняння Генрі
-рівняння Генрі
 - Коефіцієнт розподілу
- Коефіцієнт розподілу
У процесі адсорбції відбувається зміна хімічних потенціалів.
Для об'ємної фази: 
Для газу на поверхні: 
У стані рівноваги  , тобто.
, тобто.

У рівнянні Генрі константа залежить від концентрації
Рівняння Генрі виконується в області низьких тисків та концентрацій. У міру зростання концентрації можливі 2 типи відхилень від закону Генрі:
1 – позитивні відхилення, D зменшується, А зменшується
2 – негативні відхилення, D – збільшується, А – збільшується.
Тип відхилення визначається переважанням тієї чи іншої типу взаємодії адсорбент-адсорбат.
При сильному адгезійному взаємодії коефіцієнти активності зростають – позитивне відхилення. У разі когезійної взаємодії спостерігаються негативні відхилення.
Мономолекулна адсорбція.
Ізотерма Лангмюр.
Найпростіші закономірності були отримані теоретично генрі. Ленгмюр запропонував теорію, згідно з якою адсорбція розглядається як квазіхімічна реакція. При цьому:
Поверхня енергетично однорідна.
Адсорбція локалізована, кожен адсорбційний центр взаємодіє з однією молекулою адсорбату.
Молекули адсорбату не взаємодіють між собою.
Адсорбція моношарова.

 - Поверхня,
- Поверхня,  - адсорбат,
- адсорбат,  - Адсорбційний комплекс.
- Адсорбційний комплекс.
 , тоді концентрація адсорбційних місць:
, тоді концентрація адсорбційних місць:  ,
, - Гранична адсорбція.
- Гранична адсорбція.
 тоді константа реакції:
тоді константа реакції: 

 - Рівняння Лангмюра.
- Рівняння Лангмюра.
Залежність адсорбції від концентрації
1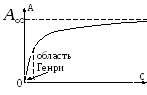 )
)
 ,
,
2) область високих концентрацій
 - гранична адсорбція, утворення мономолекулярного шару
- гранична адсорбція, утворення мономолекулярного шару
Для енергії Гіббса: .
g – ентропійний множник.
У разі ізотерми Генрі енергія Гіббса характеризує перехід адсорбату зі стандартного стану в обсязі стандартного стану на поверхні. У разі ізотерми Ленгмюра  характеризує ступінь спорідненості адсорбенту та адсорбату.
характеризує ступінь спорідненості адсорбенту та адсорбату.
 знаходять із ізабори Вант-Гоффа.
знаходять із ізабори Вант-Гоффа.

 тоді
тоді  , звідси
, звідси  .
.

 - Ступінь заповнення поверхні.
- Ступінь заповнення поверхні.




 - Число вільних місць,
- Число вільних місць,  - Число зайнятих місць.
- Число зайнятих місць.
 ,
,

Тобто. в області високих концентрацій кількість вільних місць обернено пропорційно кількості адсорбату.
Адсорбція суміші газів на однорідній поверхні.
У цьому випадку процес адсорбції розглядають як дві паралельні реакції.
 (1)
(1)

 (2)
(2)


Адсорбція суміші газів на неоднорідній поверхні.
У разі неоднорідної поверхні не можна обмежуватись середніми заповненнями.
Внаслідок конкурентної боротьби, на ділянках різних типів можлива локалізація різних адсорбатів.
В цьому випадку відношення  .
.
 ,
,
 - Тиск насиченої пари адсорбату.
- Тиск насиченої пари адсорбату.
 ,
,
 - теплоти адсорбції.
- теплоти адсорбції.
"+" - симбатна залежність, "-" - антибатна залежність, "Н" - кореляції немає.
«+» - адсорбція протікає за однаковим механізмом. На найбільш енергетично вигідних ділянках переважно адсорбується газ, що має велику спорідненість до поверхні.
«-» - адсорбція протікає за різними механізмами і до певного моменту часу конкурентної боротьби за поверхню немає.
Мономолекулярна адсорбція переважно реалізується при фізичній адсорбції газів за малих значень. p, а також на межі розділу рідина/газ.
Полімолекулярна адсорбція.
Теорія БЕТ(Брунауер, Еммет, Теллер).
У разі коли утворення моношару недостатньо для компенсації поверхневої енергії, адсорбція полімолекулярна і її можна розглядати як результат вимушеної конденсації під дією поверхневих сил.
Основні положення:
У разі потрапляння молекули адсорбату на зайняте місце утворюється кратний комплект.
У міру наближення pдо p sзменшується кількість вільних адсорбційних місць. Спочатку збільшується, та був зменшується кількість місць, зайнятих одиничними, подвійними тощо. комплектами.
При p =p s адсорбція перетворюється на конденсацію.
Горизонтальні взаємодії відсутні.
Для першого шару виконується ізотерма Лангмюр.
Поверхня сприймається як сукупність адсорбційних місць. Справедлива умова динамічної рівноваги: швидкість конденсації на вільних місцях дорівнює швидкості випаровування із зайнятих.
a – коефіцієнт конденсації (частка молекул, що сконденсувалися на поверхні);
 ,
,
Zm – максимальна кількість вільних місць.
 - частота коливань атомів у напрямі перпендикулярному до поверхні.
- частота коливань атомів у напрямі перпендикулярному до поверхні.
Для першого шару умови динамічної рівноваги:



 тоді
тоді
 - Рівняння Лангмюра.
- Рівняння Лангмюра.
Для другого шару буде справедливо: 
Для i-го шару: 
Для спрощення приймають, що і ν однакові всім шарів, крім першого. Для всіх шарів, крім першого теплота адсорбції, постійна. Для останнього шару теплота адсорбції дорівнює теплоті конденсації. В результаті було отримано рівняння
 (*)
(*)
C- Константа,
У разі теорії БЕТ, константа Зхарактеризує енергію Гіббса чистої адсорбції. Рівняння містить лише одну константу, а також це рівняння дуже важливе для визначення питомої поверхні адсорбенту.
Оскільки в результаті адсорбції теплота виділяється, визначення питомих поверхонь ведуть за низьких температур.
 ????????????
????????????


Основний недолік теорії- Нехтування горизонтальними взаємодіями на користь вертикальних.
Рівняння виконується в області значень  від 0,05 до 0,3.
від 0,05 до 0,3.
Там де  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – позначається взаємодія адсорбат – адсорбат.
> 0,3 – позначається взаємодія адсорбат – адсорбат.
Врахування взаємодій адсорбат-адсорбат.
Взаємодія виявляється при адсорбції на неполярній поверхні розгалужених молекул або молекул. Здібних утворювати асоціати. І тут змінюється форма ізотерм адсорбції.
А  дсорбент не полярний.
дсорбент не полярний.
Графіку 1 відповідають слабкі взаємодії адсорбат-адсорбат, сильне адсорбат-адсорбент.
Графіку 2 відповідають сильне взаємодія адсорбат-адсорбат, сильне адсорбат-адсорбент.
Графіку 3 відповідають сильне взаємодія адсорбат-адсорбат, слабкий адсорбат-адсорбент.
 ,
,

У разі взаємодії між молекулами адсорбату потрібний облік зміни коефіцієнтів активності. І це рівняння записують у вигляді:
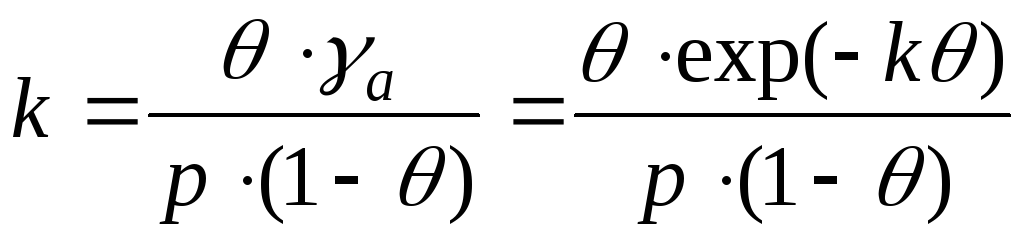 - Рівняння Фрункіна, Фаулера, Гугенгейма.
- Рівняння Фрункіна, Фаулера, Гугенгейма.
k- Атракціонна постійна.
Потенційна теорія Поляні.
Ця теорія не виводить будь-якого типу ізотерми адсорбції, а дозволяє розрахунку ізотерм при іншій температурі.
Адсорбція– це результат тяжіння адсорбату до поверхні адсорбенту за рахунок дії адсорбційного потенціалу, який не залежить від присутності інших молекул та залежить від відстані між поверхнею та молекулою адсорбату.
 ,
,
 - Адсорбційний потенціал.
- Адсорбційний потенціал.
Оскільки поверхня неоднорідна, відстань замінюють на адсорбційний об'єм.  .Адсорбційний обсяг– це обсяг, укладений між поверхнею та точкою, що відповідає даному значенню
.Адсорбційний обсяг– це обсяг, укладений між поверхнею та точкою, що відповідає даному значенню  .
.
Адсорбційний потенціал– це робота перенесення 1 моль адсорбату поза даним адсорбційним об'ємом у цю точку адсорбційного об'єму (або робота перенесення 1 моль насиченої пари адсорбату, що знаходиться в рівновазі з рідким адсорбатом без адсорбенту в рівноважну з адсорбентом парову фазу).

Характеристична крива
 - адсорбційний потенціал,
- адсорбційний потенціал, 
Для даного адсорбенту та різних адсорбатів справедливо: 
Для різних типів адсорбатів  ,
,
де  потенціали для ізотерм адсорбції при відносних тисках
потенціали для ізотерм адсорбції при відносних тисках  для адсорбату 1 та для адсорбату 2. Це відношення є величина постійна.
для адсорбату 1 та для адсорбату 2. Це відношення є величина постійна.
 - Коефіцієнт афінності
- Коефіцієнт афінності
Теорія капілярної конденсації.
Перебіг процесу адсорбції великою мірою залежить від структури пористого тіла.
|
Мікропористі | |
|
Перехіднопористі | |
|
Макропористі |
У разі мікропористих сорбентів поля адсорбційних сил перекриваються. У разі макропористих сорбентів пори виконують роль транспортних каналів. Процеси коденсації найбільш значущі у перехіднопористих тілах. Капілярна конденсація починається за певних значень pі  коли частина поверхневої енергії вже компенсована. Необхідна умова – поверхня має бути самчується. Процеє описується рівнянням Томпсона – Кельвіна.
коли частина поверхневої енергії вже компенсована. Необхідна умова – поверхня має бути самчується. Процеє описується рівнянням Томпсона – Кельвіна.

 - для випадку змочування центр кривизни знаходиться в газовій фазі.
- для випадку змочування центр кривизни знаходиться в газовій фазі.
У разі капілярної конденсації ізотерму адсорбції має гістерезисний вигляд. Процес адсорбції відповідає нижня гілка, процесу десорбції - верхня.
Всі види пір можна звести до трьох видів:
|
Конічні |
Циліндричні з одним закритим кінцем |
Циліндричні з двома відкритими кінцями |
|
Процесне наповнення здійснюється з дна пори. Ізтерма адсорбції та ізотерма десорбції в цьому випадку збігаються, оскільки процес адсорбції починається зі сфери та процес десорбції також починається зі зникнення деяких сфер.
↓ |
Гістерези немає. Прямий та зворотний хід описуються рівнянням:
|
Дна ніде немає, заповнення пори піде на стінки циліндра.
циліндр: Ізотерма і матиме гістерезисний вигляд.
↓ |


 - сфера,
- сфера, ,
,



В  умовах змочування конденсація здійснюється за нижчих тисків, що енергетично вигідно. По десорбційної гілки одержують криві розподілу пір за розмірами.
умовах змочування конденсація здійснюється за нижчих тисків, що енергетично вигідно. По десорбційної гілки одержують криві розподілу пір за розмірами.
Максимум диференціальної кривої зміщений вліво щодо точки інтегральної перегину. Загальний обсяг малих пір невеликий, проте має великі значення площі поверхні. Зі збільшенням розміру доби, їх обсяг зростає як  , а площа як
, а площа як  , за рахунок цього і спостерігається усунення максимуму диференціальної кривої.
, за рахунок цього і спостерігається усунення максимуму диференціальної кривої.
Адсорбція на межі твердого тіла – рідина.
У разі адсорбції на кордоні тверде тіло – газ, ми нехтували одним компонентом. У разі адсорбції на межі тверде тіло – рідина адсорбат витісняє з поверхні адсорбенту молекули розчинника.
 ,
,
Справедливе рівняння:

 ,
,
N 1 , N 2 – мольні частки розчинника та компонента, N 1 + N 2 = 1, тоді
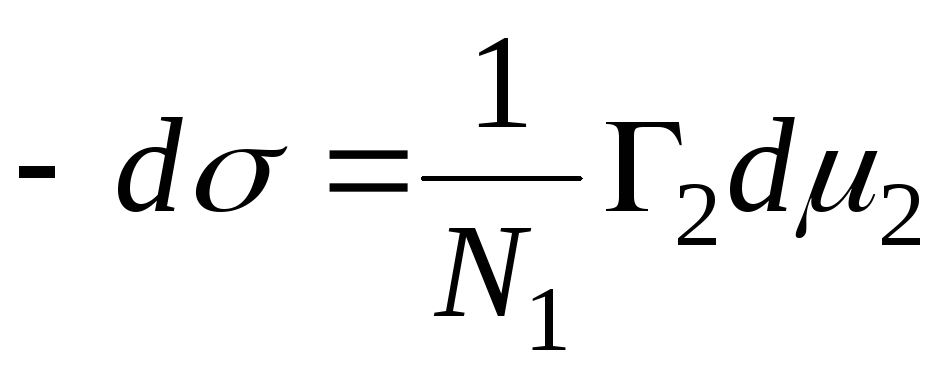 ,
=>
,
=>
 , Тоді- рівняння адсорбції для межі розділу фаз тверде тіло - рідина.
, Тоді- рівняння адсорбції для межі розділу фаз тверде тіло - рідина.
Адсорбція (Г) > 0 при  <
0
<
0
Якщо значення  для компонента та розчинника сильно різні, у цьому випадку залежність Гвід Nмає екстремум при значенні N
~ 0,5.
для компонента та розчинника сильно різні, у цьому випадку залежність Гвід Nмає екстремум при значенні N
~ 0,5.
Е  слі
слі  мають близькі значення, у разі можлива зміна знака адсорбції. Залежність Гвід Nперетинає вісь абсцис
мають близькі значення, у разі можлива зміна знака адсорбції. Залежність Гвід Nперетинає вісь абсцис
Точка перетину функції Г(N) з віссю абсцис називається адсорбційним азеотропом. Це означає, що два компоненти не можуть бути поділені на даному адсорбенті.
Рівняння ізотерми адсорбції із константою обміну.
При адсорбції на межі тверде тіло – рідина постійно відбувається перерозподіл компонентів між поверхнею адсорбенту та об'ємом розчину.
 - компоненти (- - відносяться до поверхні)
- компоненти (- - відносяться до поверхні)

 ,
,
 ,
, .
.
 ,
,


Адсорбція на межі рідина-газ
Р  Розглянемо зміну концентраційного профілю в міру перетину межі розділу рідина-газ. Нехай компонент 2 лет.
Розглянемо зміну концентраційного профілю в міру перетину межі розділу рідина-газ. Нехай компонент 2 лет.
Cs – концентрація у поверхневому шарі.
Виходячи з визначення надмірної адсорбції

Якщо компонент не леткий, то величина адсорбції запишеться так:
П  рі
рі 

У рівнянні  природа речовини описується похідною
природа речовини описується похідною  .
.
Ізотерма поверхневого натягу може мати вигляд 1 або 2:
1 – поверхнево-інактивні речовини
2 – поверхнево-активні речовини
Поверхневою активністю g називається здатність речовин знижувати поверхневий натяг у системі.

 - Товщина поверхневого шару
- Товщина поверхневого шару
C s- Концентрація компонента в поверхневому шарі
З– об'ємна концентрація
Для гомологічного ряду існує правило:
 - правило Траубо Дюкло
- правило Траубо Дюкло
Для гомологічного ряду ізотерму адсорбції виглядає так:
Замість A пишемо Р, оскільки адсорбція надлишкова у поверхневому шарі.
Ізотерма поверхневого натягу:

 - Поверхневий натяг чистого розчинника.
- Поверхневий натяг чистого розчинника.
 - фундаментальне адсорбційне рівняння;
- фундаментальне адсорбційне рівняння;
 - Рівняння Лангмюра.
- Рівняння Лангмюра.
Вирішимо їх спільно:

- рівняння Шишковського.
B- Константа для гомологічного ряду.
A- при переході від одного гомолога до іншого збільшується у 3-3,5 рази 
![]()
1 – область малих концентрацій
![]()
2 – середня концентрація
3 – мономолекулярний шар
Поверхневоактивні речовини є дифільні молекули, тобто. включають полярну групу та неполярний вуглеводневий радикал.
o – полярна частина молекули.
| - Неполярна частина молекули.
У полярному розчиннику молекули ПАР орієнтуються таким чином, що полярна частина молекули звернена до розчинника, а неполярна виштовхується у газову фазу.
У рівнянні Шишковського  , вона стала для гомологічного ряду.
, вона стала для гомологічного ряду.
Поверхнево-активна дія починає проявлятися з n>5. При концентраціях більших, ніж концентрація мономолекулярного шару, в розчинах ПАР відбувається міцелоутворення.
Міцелла– називається агрегат молекул дифільних ПАР, вуглеводневі радикали яких утворюють ядро, а полярні групи звернені у водну фазу.
Маса міцели – міцеляльна маса.
Ч  сло молекул – число агрегації.
сло молекул – число агрегації.
Сферичні міцели
У разі міцелоутворення в розчині встановлюється рівновага
ККМ – критична концентрація міцелоутворення.
Оскільки ми вважаємо міцеллу окремою фазою:
Для гомологічного ряду існує емпіричне рівняння:
a- Енергія розчинення функціональної групи.
b – інкремент адсорбційного потенціалу, робота адсорбції на одну метиленову ланку.
– інкремент адсорбційного потенціалу, робота адсорбції на одну метиленову ланку.
Наявність у міцелах вуглеводневого ядра створює можливість для розчинення у водних розчинах ПАР сполук, які не розчиняються у воді, це явище називається солюбілізацією (те, що розчиняється – солюбілізат, ПАР – солюбілізатор).
Бруд може бути зовсім не полярний, може містити як полярну, так і не полярну частину і орієнтуватиметься як молекула ПАР.
У будь-якому разі при солюбілізації відбувається збільшення міцелярної маси та числа агрегації не тільки за рахунок включення солюбілізату, але й за рахунок збільшення числа молекул ПАР, необхідних підтримки рівноважного стану.
Солюбілізація тим ефективніше, що менше молекулярна маса солюбілізату.

 ~ 72 мН\м.
~ 72 мН\м.
 ~ 33 мН\м.
~ 33 мН\м.
Ефективність ПАР залежить від величини ККМ.
Двовимірний тиск поверхневого шару

→ сили поверхневого натягу.
- двомірний тиск.
Поверхневий шар - це сила, що дорівнює різниці поверхневих натягів розчину ПАР і чистого розчинника, спрямована в бік чистої поверхні.
Між розчином та поверхневим шаром встановлюється рівновага



При  існує область, де
існує область, де  лінійно залежить від концентрації.
лінійно залежить від концентрації.
Г [моль/м2].
 -Площа, займана одним молем речовини
-Площа, займана одним молем речовини
Тоді ізотерма двомірного тиску матиме вигляд 
 - Ізотерма двомірного тиску.
- Ізотерма двомірного тиску.
Залежність  від S М:
від S М:

При  - Двовимірний тиск різко зростає. При
- Двовимірний тиск різко зростає. При  двовимірний деформується, що викликає різке зростання
двовимірний деформується, що викликає різке зростання  .
.
Плівка з обох боків обмежена однаковими фазами називається двосторонньою. У таких плівках спостерігається постійний рух маткового розчину.
Плівки завтовшки менше 5 нм називаються чорними плівками.

Адсорбційні шари повинні мати дві характеристики: в'язкість і легкорухливість, плинність і пружність.
Ефект Марангоні – це самозалікування.
Трикутник Гіббса,  - надлишковий тиск.
- надлишковий тиск.
Плівка розтягнулася і за рахунок того, що частина рідини пішла, ПАР усуваються у вільне місце. Трикутник Гіббса.
Ефект адсорбційної міцності тел.
На поверхні плівки завжди існує адсорбційний шар, для якого тоді
Рівняння Лангмюра:


 у двомірний тиск
у двомірний тиск
 - аналог рівняння Шишковського
- аналог рівняння Шишковського
Електрокінетичні явища. Подвійний електричний прошарок (ДЕС).
Модель Гелемгольця. Теорія Гуї-Чапмена.
1808 р. Рейс

U – образна трубка, занурюють у неї 2 електроди. Закон сполучених судин порушується і відбувається зміна рівня рідини у трубці – електрокінетичні явища.
Кінетичні явища:
Електрофорез
Електроосмос
Потенціал течії (протікання)
Потенціал седиментації
1 і 2 виникають при накладенні різниці потенціалів, 3 і 4 продавлювання та седиментація колоїдних частинок викликають появу різниці потенціалів.
Електроосмосом називається рух дисперсійного середовища щодо нерухомої дисперсної фази під дією електричного струму.
Електрофорез – це рух частинок дисперсної фази щодо нерухомого дисперсійного середовища під дією електричного струму.
П  ричина виникнення електрокінетичних явищ – це просторовий поділ зарядів та виникнення подвійного електричного шару.
ричина виникнення електрокінетичних явищ – це просторовий поділ зарядів та виникнення подвійного електричного шару.
Подвійний електричний шар є плоским конденсатором, одна обкладка утворена потенціалорізальними іонами, інша - протиіноами. Іони заражені також як потенціалозначальні ко-іони відтіснені в об'єм розчину. Відстань між обкладками  . Потенціал падає лінійно, різниця потенціалів
. Потенціал падає лінійно, різниця потенціалів  .
.
Зовнішня різниця потенціалів викликає появу модуля зсуву  - це пара сил віднесених до одиниці площі, що діють уздовж поверхні твердого тіла.
- це пара сил віднесених до одиниці площі, що діють уздовж поверхні твердого тіла.

У стані рівноваги модуль зсуву дорівнює модулю в'язкого тертя (  ).
).

У наших умовах  ,
,

 - рівняння Гелемгольця-Смалуковського
- рівняння Гелемгольця-Смалуковського
 - Лінійна швидкість зсуву я фаз.
- Лінійна швидкість зсуву я фаз.
E- Напруженість електричного поля.
 - Різниця потенціалів між обкладками
- Різниця потенціалів між обкладками
 - електрофоретична рухливість [м2/(В * с)].
- електрофоретична рухливість [м2/(В * с)].
У моделі Гелемгольця не враховується тепловий рух молекул. Реально розподіл іонів у подвійному шарі має більш складний характер.
Гуї та Чапман виділили такі причини виникнення ДЕС:
Перехід іона з однієї фази до іншої при встановленні рівноваги.
Іонізація речовини твердої фази.
Добудовує поверхню іонами, присутніми в дисперсійному середовищі.
Поляризація від зовнішнього джерела струму.
Подвійний електричний шар має розмиту чи дифузну будову. Іони прагнуть поступово розподілитися у всьому дифузному шарі.
Дифузний шар складається з протиінон, протяжність шару визначається їх кінетичною енергією. При температурі протиіної, що прагне до абсолютного нуля, максимально наближені до потенціаловизначальних іонів.
Даня теорія базується на двох рівняннях:
рівняння Больцмана

 - робота проти сил електростатичної взаємодії.
- робота проти сил електростатичної взаємодії.
 - Об'ємна щільність заряду.
- Об'ємна щільність заряду.

Рівняння Пуассона 
Оскільки товщина ДЕС багато менша за розміри частинки і для плоского ДЕС похідна за координатами  і
і  скасовується.
скасовується. 
Для е у при у<<1 функцию можно разложить в ряд Маклорена:

Обмежимося двома членами ряду, тоді:


 - Товщина ДЕС - це відстань, на якій потенціал ДЕС зменшується в eразів.
- Товщина ДЕС - це відстань, на якій потенціал ДЕС зменшується в eразів.
Чим менша температура, тим менше  . При Т→0 – плаский ДЕС. Чим більша концентрація, тим більше I, тим менше
. При Т→0 – плаский ДЕС. Чим більша концентрація, тим більше I, тим менше  .
.





 "-" означає, що потенціал з відстанню зменшується. =>
"-" означає, що потенціал з відстанню зменшується. =>
=>

 ,
,
 - Потенціал експоненційно зменшується.
- Потенціал експоненційно зменшується.
Потенціал для поверхневої щільності заряду: 
Поверхневий заряд – об'ємний заряд із протилежним знаком, проінтегрований на відстані.




=>


Там, де потенціал зменшується у 2,7 рази - 
Ємність подвійного шару 
Недолік теорії – не враховується наявність шару Гелемгольця, тобто. не враховує  звідси помилки у визначенні основних параметрів. Також не пояснює впливу іонів різної природи на товщину подвійного електричного шару.
звідси помилки у визначенні основних параметрів. Також не пояснює впливу іонів різної природи на товщину подвійного електричного шару.
Теорія Штерна. Будова колоїдної міцели.
Подвійний електричний шар складається з двох частин: щільної та дифузної. Щільний шар утворюється в результаті взаємодії потенціалутворюючих іонів зі специфічно адсорбуються. Ці іони, як правило, частково або повністю дегідратовані і можуть мати як однаковий, так і протилежний до іонів потенційно заряд. Це залежить від співвідношення енергії електростатичної взаємодії  та потенціалу специфічної адсорбції
та потенціалу специфічної адсорбції  . Іони щільного шару закріплені. Інша частина іонів розташована в дифузному шарі, ці іони вільні і можуть переміщатися углиб розчину, тобто. з області більшої концентрації в область меншої. Загальна щільність заряду складається із двох частин.
. Іони щільного шару закріплені. Інша частина іонів розташована в дифузному шарі, ці іони вільні і можуть переміщатися углиб розчину, тобто. з області більшої концентрації в область меншої. Загальна щільність заряду складається із двох частин.

 -заряд шару Гельмгольця
-заряд шару Гельмгольця
 -Заряд дифузного шару
-Заряд дифузного шару
Поверхня має кілька адсорбційних центрів, кожен із яких взаємодіє з одним противоионом. Константа такої квазіхімічної реакції дорівнює:

 , де
, де  - мольна частка протиіонів у розчині
- мольна частка протиіонів у розчині
Розподіл Гельмгольця
Потенціал зменшується лінійно
Розподіл потенціалу по Гуї. Щільного шару немає, потенціал зменшується експоненційно зі значення 
Розподіл по Штерну.
Спочатку зниження потенціалу лінійне, та був експоненціально.
При накладенні електричного поля у разі електрофорезу рухається не безпосередньо частка твердої фази, а частка твердої фази з шаром іонів, що її оточують. ДЕС повторює форму частинки дисперсної фази. При накладенні потенціалу відривається частина дифузного шару. Лінія розриву називається кордоном ковзання.
Потенціал, що виникає на межі ковзання внаслідок відриву частини дифузного шару, називається електрокінетичним потенціалом(Дзета потенціал  ).
).
Частка дисперсної фази, з навколишнім шаром протиіонів і подвійним електричним шаром називається міцеллою.
Правила написання колоїдних міцел:


1-1 зарядний електроліт
T – частка дисперсної фази.
AA – межа щільної та дифузної частини.
BB – межа ковзання.
Кордон ковзання може збігатися з лінією AA, а може й не збігатися.
Значення pH, при якому дзета-потенціал дорівнює нулю, називається ізоелектричною точкою.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. У надлишку CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m·nCa 2+ 2( n - x)Cl - ) 2 x + x Cl - - запис міцели.
CaSO 4 m – агрегат.
CaSO 4 m·nCa 2+ – ядро.
CaSO 4 m·nCa 2+ 2( n - x)Cl - - частка.
2. У надлишку Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - міцела
CaSO 4 m – агрегат.
CaSO 4 m∙nSO 4 2 + – ядро.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - частка
Рівняння Гелемгольця-Смолуховського

 - Лінійна швидкість зміщення кордонів (в електроосмосі).
- Лінійна швидкість зміщення кордонів (в електроосмосі).
 - Різниця потенціалів на обкладках конденсатора (в електроосмосі).
- Різниця потенціалів на обкладках конденсатора (в електроосмосі).


 - об'ємна швидкість перебігу розчину, S- Площа поперечного перерізу комірки.
- об'ємна швидкість перебігу розчину, S- Площа поперечного перерізу комірки.

E- Напруженість електричного поля.


 (Для електроосмосу).
(Для електроосмосу).
Для потенціалу течії:

 - потенціал
- потенціал
 - тиск на мембрану
- тиск на мембрану
Як правило, значення електрофоретичних рухливостей та електроосмотичних рухомостей менше за розрахункові. Це відбувається внаслідок:
Релаксаційного ефекту (під час руху частки дисперсної фази порушується симетрія іонної атмосфери).
Електрофоретичного гальмування (виникнення додаткового тертя внаслідок руху протиіонів).
Спотворення ліній струму у разі електропровідних частинок.
Зв'язок поверхневого натягу з потенціалом. Рівняння Ліппмана.
Утворення ДЕС відбувається мимоволі через прагнення системи знизити свою поверхневу енергію. В умовах сталості Tі pузагальнене рівняння першого та другого законів термодинаміки виглядає:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1-е рівняння Ліпмана.
- 1-е рівняння Ліпмана.
 - Поверхнева щільність заряду.
- Поверхнева щільність заряду.
 - Диференційна ємність.
- Диференційна ємність.
 - 2-е рівняння Ліпмана.
- 2-е рівняння Ліпмана.
З- Місткість.
Розв'яжемо 1-е рівняння Ліппмана та фундаментальне рівняння адсорбції:
 ,
,


 тоді
тоді
 - Рівняння Нернста
- Рівняння Нернста
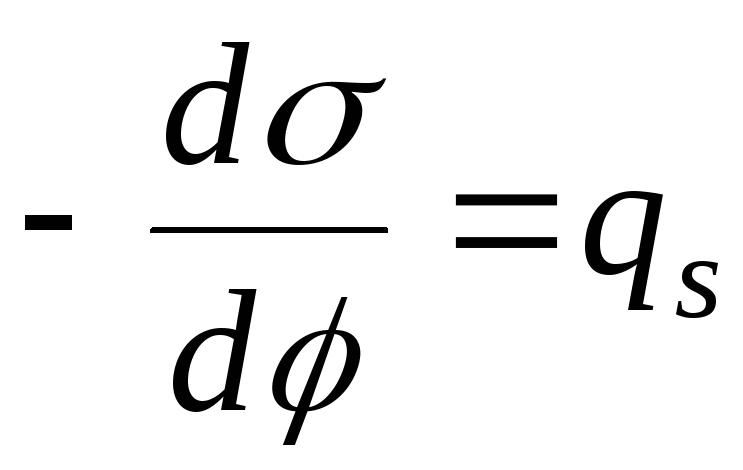 ,
,
 ,
,




 - Рівняння електрокапілярної кривої (ЕКК).
- Рівняння електрокапілярної кривої (ЕКК).
В  :
: , але
, але 
Катіонні ПАР (КПАВ) знижують катодну гілка ЕКК.
Аніонні ПАР (АПАВ) знижують анодну гілку ЕКК.
Неіоногенні ПАР (НПАВ) знижують середню частину ЕКК.
Стійкість дисперсних систем. Розклинює тиск.
Дисперсні системи можна поділити:
Системи нестійкі термодинамічно можуть бути кінетично стійкі за рахунок переходу в метастабільний стан.
Розрізняють два види стійкості:
Седиментаційна стійкість (стосовно силі тяжкості).
Агрегативна стійкість. (по відношенню до злипання)
Коагуляція- Це процес злипання частинок, що призводить до втрати агрегатної стійкості. Коагуляцію може спричинити зміну температури, pH, перемішування, ультразвук.
Розрізняють коагуляцію:
Оборотна.
Необоротна.
Коагуляція протікає під час введення електролітів.
Правила коагуляції:

Плівка– це частина системи, що перебуває між двома міжфазними поверхнями.
тиск, що розклинюєвиникає при різкому зменшенні товщини плівки в результаті взаємодії поверхневих шарів, що зближуються.

«-» - при зменшенні товщини плівки тиск, що розклинює, зростає.
P 0 - Тиск в об'ємній фазі, яка є продовженням прошарку.
P 1 – тиск у плівці.
Теорія сталості. ДЛФО (Дерягін, Ландау, Фервей, Овербек).
Відповідно до теорії ДЛФО у розклинювальному тиску виділяють дві складові:
ЕлектростатичнаП Е (позитивна, вона обумовлена силами електростатичного відштовхування). Відповідає зменшенню енергії Гіббса у разі зростання товщини плівки.
МолекулярнаП М (негативна, обумовлена дією сил тяжіння). Обумовлена стиском плівки за рахунок хімічних поверхневих сил, радіус дії сил десяті долі нм з енергією близько 400 кДж/моль.
Повна енергія взаємодії:

 - система агрегативно стійка
- система агрегативно стійка
 - нестійка система
- нестійка система
П  оложна складова.
оложна складова.
Збільшення обумовлено збільшенням потенційної енергії при стисканні тонких плівок. Для плівок великої товщини надмірна енергія іонів скомпенсована і дорівнює енергетичній взаємодії в обсязі дисперсійного середовища.
Якщо  (
( - Товщина плівки,
- Товщина плівки,  - радіус іона) утончення плівки призводить до зникнення та зменшення в ній молекул та іонів з мінімальною поверхневою енергією. Число сусідніх частинок зменшується, внаслідок чого потенційна енергія частинок, що залишилися в плівці, зростає.
- радіус іона) утончення плівки призводить до зникнення та зменшення в ній молекул та іонів з мінімальною поверхневою енергією. Число сусідніх частинок зменшується, внаслідок чого потенційна енергія частинок, що залишилися в плівці, зростає.


Теорія ДЛФО взаємодію частинок розглядає як взаємодію пластин.

Частинки не взаємодіють



 - рівняння Лапласа,
- рівняння Лапласа,  ,
,


Для слабко заряджених поверхонь
Для сильно заряджених поверхонь:

Молекулярна складова – взаємодія двох атомів:
 ~
~
Взаємодія атома з поверхнею:

Візьмемо дві платівки:
Д  ля отримання молекулярної складової необхідно провести підсумовування всіх енергій взаємодії атомів правої та лівої пластин.
ля отримання молекулярної складової необхідно провести підсумовування всіх енергій взаємодії атомів правої та лівої пластин.
де  - постійна Гамакера (враховує природу тіл, що взаємодіють).
- постійна Гамакера (враховує природу тіл, що взаємодіють).
Т.ч. Енергія взаємодії частинок у системі може бути виражена за допомогою потенційних кривих.
I – первинний потенційний мінімум. Це зона незворотної коагуляції, сили тяжіння переважають.
II – зона агрегативної стійкості, що переважають сили відштовхування.
ІІІ – вторинний потенційний мінімум (або зона флокуляції). Між частинками дисперсної фази існує прошарок електроліту, і частинки можуть бути розділені та переведені в зону агрегативної стійкості.

Крива 1 – система агрегативно стійка.
Крива 2 – у зоні I стійка, у зоні II не стійка.
Крива 3 – у системі відбулася коагуляція.
Крива 4 – у точці 4 сумарна енергія взаємодії U=0,  ця точка екстремуму відповідає початку швидкої коагуляції.
ця точка екстремуму відповідає початку швидкої коагуляції.
Існує два випадки:
1. Поверхні слабозаряджені:
U = U Е + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - це товщина прошарку, що відповідає початку процесу коагуляції.
- це товщина прошарку, що відповідає початку процесу коагуляції.






 - для слабко заряджених поверхонь
- для слабко заряджених поверхонь
 тоді
тоді 
2. Для сильнозаряджених поверхонь:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Зводимо (3) у квадрат

Коагуляція:
При специфічній адсорбції іони можуть адсорбуватись у надеквівалентній кількості таким чином, що поверхня може змінити свій заряд. Відбувається перезаряджання поверхні.
У разі специфічної адсорбції можуть адсорбуватися іони як протилежних знаків, а й одного.
Якщо адсорбуються іони того ж знака, що й поверхня, то поверхневому шарі відбуватиметься не падіння потенціалу, а його зростання.

Нейтралізаційна коагуляція (протікає за участю слабозаряджених частинок і залежить не тільки від заряду електроліту-коагулятора, а й від потенціалу на межі щільного та дифузного шару).
Теорія швидкої коагуляції Смолухівського.

Залежність швидкості коагуляції від концентрації електроліту.
I – швидкість коагуляції мала,
II – швидкість коагуляції практично пропорційна концентрації електроліту.
III – область швидкої коагуляції, швидкість майже залежить від концентрації.
Основні положення:
Вихідна золь монодисперсна, подібні частинки мають сферичну форму.
Усі зіткнення часток результативні.
При зіткненні двох первинних частинок утворюється вторинна. Вторинна + первинна = третинна. Первинне, вторинне, третинне – кратність.
У термінах хімічної кінетики процес коагуляції може бути описаний рівнянням:

Рішенням буде рівняння: 
 - Час половинної коагуляції. Це час, протягом якого кількість частинок золю зменшується вдвічі.
- Час половинної коагуляції. Це час, протягом якого кількість частинок золю зменшується вдвічі.

 ,
,
 ,
,
 ,
,


У міру збільшення кратності максимум кривих коагуляції зсувається у бік більших значень  .
.
Недоліки:
Припущення про монодисперсність.
Припущення результативності всіх зіткнень.
Рекомендуємо також
 Імпульсний блок живлення: ремонт та доробка
Імпульсний блок живлення: ремонт та доробка
 Дистанційне керування світлом
Дистанційне керування світлом
 Навчання плавання дітей дошкільного віку
Навчання плавання дітей дошкільного віку
 Нотатки для майстра - домашні побутові сигналізатори
Нотатки для майстра - домашні побутові сигналізатори
 Годинник пропелер на Atmega8
Годинник пропелер на Atmega8
 Пристрій та приклади застосування реле, як вибрати та правильно підключити реле Мікроконтролер та реле найпростіші схеми включення
Пристрій та приклади застосування реле, як вибрати та правильно підключити реле Мікроконтролер та реле найпростіші схеми включення