Termodynamik för adsorptionsprocessen. Termodynamik för adsorptionsprocesser Förändringar i termodynamiska funktioner under adsorption
Adsorption som en spontan koncentration av molekyler på ytan åtföljs av en minskning av systemets entropi. Eftersom kriteriet för processens spontanitet är
∆N - T · ∆S = ∆G< 0,
då är adsorption endast möjlig vid ∆Н< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. Med en ökning av temperaturen skiftar jämvikten mot den endotermiska processen, d.v.s. desorption.
Adsorption på fast yta
1. monomolekylär adsorption.
Enligt Langmuirs teori interagerar adsorbatmolekyler med den adsorberande ytan och bildar så småningom ett monomolekylärt skikt. I detta fall, graden av fyllning () av ytan med det adsorberade ämnet under adsorption från gasfasen
från vätska
där K är jämviktskonstanten (adsorptionskonstanten);
p är partialtrycket för den adsorberade gasen;
c är koncentrationen av det adsorberade ämnet.
Beroendet av β på p (eller c) representeras av en graf (adsorptionsisoterm, Т = const) i fig. 1. 1.3.

Ris. 1.3. Fyllnadsgrad av ytan med adsorberat ämne
Vid låga koncentrationer och partialtryck är adsorptionen proportionell mot koncentrationen eller partialtrycket:
R<< 1, β ≈ К· r oris<< 1, β ≈ К· si e. den initiala delen av isotermen är ungefär linjär och tan α \u003d K (tg α bestäms av kurvans lutning vid p (eller c) → 0: eller ).
Om - antalet mol av det adsorberade ämnet per 1 g av adsorbenten; - det högsta möjliga antalet mol av det adsorberade ämnet per 1 g av adsorbenten ("monolagerkapacitet"), sedan
Ersätter β i ekvation (1.3) (för fallet med adsorption från gasfasen, koncentrationen med i ekvationerna bör ersättas med tryck R), vi får:
 (1.6)
(1.6)
Eftersom och K i detta par av adsorbent-adsorbent är konstanter (vid T= const), då kan beroendet hittas och Till(Fig. 1.4).

Ris. 1.4. Grafisk lösning av adsorptionsekvationen
erhålls genom att extrapolera det experimentella linjära beroendet till () = 0; och sedan , då .
Värdet kan användas för att bestämma adsorbentens specifika yta UD (i m 2 per 1 g adsorbent), om arean ω som upptas på ytan av en molekyl av adsorbenten är känd (bestäms utifrån molekylens storlek):
UD = · ω · Na, (1,7)
där Na är Avogadros tal (Na = 6,02 10 23).
I sin tur kan det kända värdet för SD användas för att beräkna eller ω av vilken substans som helst genom dess adsorption på en given adsorbent.
2. Polymolekylär adsorption.
Ekvation (1.5) beskriver en kurva med mättnad, dvs. på
p (eller c) → ∞ tenderar mot gränsvärdet lika med (Fig. 1.5, a).

Fig.1.5. Adsorptionsisotermer:
a – adsorption med mättnad; b – polymolekylär adsorption
Men i vissa fall ser adsorptionsisotermer ut som de som visas i fig. 1,5b, dvs. når inte gränsen ens vid hög p (eller c).
Beroenden av den typ som visas i fig. 1,5b motsvarar polymolekylär adsorption. Som regel är sådana isotermer karakteristiska för ämnen med starka intermolekylära interaktioner (till exempel vatten). När adsorptionscentra på ytan av adsorbenten är upptagna (det monomolekylära lagret är mättat) sker "landningen" av nästa adsorbatmolekyler på grund av intermolekylära interaktioner med redan adsorberade molekyler (Fig. 1.6). Värmen av sådan adsorption är nära i absolut värde, men motsatt i tecken, till värmen från avdunstning av motsvarande vätska (tänk varför).

Fig.1.6. Adsorptionsschema:
a - monomolekylär adsorption; b - polymolekylär adsorption
När du kommer närmare R till det mättade ångtrycket hos det adsorberade ämnet börjar det kondensera på ytan av adsorbenten; som ett resultat ökar det snabbt med ökande R.
Termodynamik för adsorptionsprocesser.
| Parameternamn | Menande |
| Artikelns ämne: | Termodynamik för adsorptionsprocesser. |
| Rubrik (tematisk kategori) | Utbildning |
Grundläggande definitioner och metoder för att klassificera adsorptionsprocesser.
Adsorption avser fenomen som uppstår som ett resultat av en spontan minskning av ytenergi.
Adsorption- Processen för spontan reversibel eller irreversibel omfördelning av komponenterna i ett heterogent system mellan ytskiktet och volymen av en homogen fas.
I flerkomponentsystem är den komponent som sänker gränsytspänningen att föredra framför ytskiktet. I enkomponentsystem förändras dess struktur under bildandet av ytskiktet (en viss orientering av atomer och molekyler, polarisering), kallad autoadsorption.
Den tätare fasen på vilken adsorptionsinteraktioner är lokaliserade kallas adsorbent. Ämnet som omfördelas mellan volymen av den homogena fasen och ytskiktet betecknas med termen '' adsorbatʼʼ.
I vissa fall är adsorptionsprocessen reversibel. I detta fall, under vissa förhållanden, kan en del av de adsorberade molekylerna, som ett resultat av molekylära kinetiska fenomen, passera från ytskiktet in i fasens volym. Den omvända processen för adsorption kallas desorption.
Metoder för att klassificera adsorptionsprocesser.
Klassificering av adsorptionsprocesser enligt aggregationsstadiet för de interagerande faserna. Med hänsyn till beroendet av det aggregerade tillståndet för intilliggande faser, särskiljs följande typer av adsorptionsprocesser:
Adsorption av gaser på fasta adsorbenter;
Adsorption av lösta ämnen vid gränssnitten ʼʼfast-vätskaʼʼ och ʼʼvätska-vätskaʼʼ;
Adsorption av ytaktiva ämnen vid gränssnittet ''vätskegas''.
Klassificering av adsorptionsprocesser enligt mekanismen för interaktion mellan adsorbenten och adsorbatet. Adsorption kan betraktas som interaktionen mellan adsorbatmolekyler och adsorbentens aktiva centra. Enligt mekanismen för deras interaktion är följande typer av adsorption uppdelade:
1) fysisk (molekylär) adsorption- interaktionen mellan adsorbatets molekyler och adsorbenten utförs på grund av van der Waals-krafterna, vätebindningar (utan förekomsten av kemiska reaktioner);
2) kemisk adsorption (kemisorption)- fästningen av adsorbatmolekyler till adsorbentens aktiva centra sker som ett resultat av kemiska reaktioner av olika typer (med undantag för jonbytesreaktioner);
3) jonbytaradsorption (jonbyte) - omfördelningen av adsorbatsubstansen mellan lösningen och den fasta fasen (jonbytaren) enligt mekanismen för jonbytesreaktioner.
För en kvantitativ beskrivning av adsorptionsprocesser används två kvantiteter.
1) Absolut adsorptionär mängden (mol) eller massan (kg) av adsorbatet per ytenhet eller massa av adsorbenten. Beteckning - A; enhet: mol/m 2 , mol/kg, kg/m 2 , kg/kᴦ.
2) Gibbs (överskott) adsorption- ett överskott av adsorbatämne i ett ytskikt av en viss tjocklek jämfört med dess mängd i volymen av en homogen fas, hänfört till en enhetsyta eller massa av adsorbenten. Beteckning - G; enhet: mol/m 2 , mol/kᴦ.
Förhållandet mellan absolut och överskottsadsorption kan illustreras med hjälp av ekvationen:
G \u003d A - c * h (3,1)
där c är ämnets jämviktskoncentration i fasens volym, mol/m3;
h är tjockleken på ytskiktet, villkorligt taget lika med 10 -9 m.
I flerkomponents heterogena system, när en eller annan komponent omfördelas mellan volymen av en homogen fas och ytskiktet, är ekvationen för ytans överskott av inre energi giltig:
U = T * S + s * s + Sm i * n i (3,2)
Genom att föra alla termer i ekvationen till enhetsarean för gränssnittsytan får vi:
U s = T * S s + s + Sm i * Г i (3.3)
där Г i = n i/s är överskottet av den i:te komponenten i ytskiktet, det vill säga Gibbs adsorption.
För ett enkomponentsystem har ekvation (3.3) formen:
G s = s + m * Г (3,4)
där G s = U s - T * S s är Gibbs-energin på ytan eller arbetet med att skapa en enhetsarea av ytan;
m * Г - komprimering av det adsorberade ämnets substans i ytskiktet.
Utifrån ekvation (3.4) kan vi dra slutsatsen att under adsorptionen består arbetet med att skapa en gränsyta av arbetet med att bilda en yta (bryta kohesiva bindningar i huvuddelen av adsorbatfasen) och komprimera ämnet i ytskiktet.
I ett tillstånd av dynamisk jämvikt mellan adsorbenten och adsorbatet, förändringen i Gibbs-energin för ett heterogent system ΔG = 0, beskrivs termodynamiken för adsorptionsprocessen med en ekvation som kallas Gibbs fundamentala adsorptionsekvation:
Ds = SГ i * dm i (3,5)
Denna ekvation är universell, eftersom den är giltig för alla typer av adsorptionsprocesser
Särskilda fall av Gibbs adsorptionsekvation.
1) Adsorption från lösningar.
För den kemiska potentialen för den i:te komponenten i systemet under adsorption vid gränssnitten "vätska - fast adsorbent" och "vätska - gas" är ekvationerna giltiga:
m i = m i 0 + R*T*ln a i (3,6)
dm i = R*T* d ln a i (3,7)
där m i 0 är den kemiska potentialen för den i:te komponenten i systemet under standardförhållanden;
a i – aktivitet för den i:te komponenten i systemet under standardförhållanden.
Baserat på detta kommer Gibbs adsorptionsekvation att ta formen:
Г i = - a i / R*T * (ds / da i) (3.8)
För icke-elektrolytlösningar tar vi en i \u003d c i, sedan:
Г i \u003d - s / R * T * (ds / ds) (3.9)
För elektrolytlösningar:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
där c ± är den genomsnittliga jonkoncentrationen av lösningen;
n är den stökiometriska koefficienten.
2) Adsorption av ämnen från gasfasen.
I enlighet med Mendel-eev-Claiperons ekvation:
P \u003d c * R * T (3.11)
I detta avseende är Gibbs ekvation för adsorption av gaser på fasta adsorbenter skriven i följande form:
Г i = - Р / R*T * (ds / dР) (3.12)
I praktiken tillåter Gibbs adsorptionsekvation, enligt mätning av ytspänning vid olika värden av vätskekoncentration eller jämviktsgastryck, att beräkna mängden adsorption av ämnen i gränsskiktet, för vilka ytspänningen bestäms.
Termodynamik för adsorptionsprocesser. - koncept och typer. Klassificering och funktioner i kategorin "Termodynamik för adsorptionsprocesser." 2017, 2018.
Aktuell sida: 6 (boken har totalt 19 sidor) [tillgängligt läsutdrag: 13 sidor]
Font:
100% +
34. Typ av adsorptionskrafter
Interaktionen mellan adsorbentens molekyler med adsorbentens yta vid den sk. fysisk adsorption kan bero på olika orsaker. Då kan potentialen som bestämmer interaktionen mellan en adsorbentmolekyl och en atom i ett opolärt adsorptivt uttryckas på följande sätt:
θ = −Cr 6 +Br 12 ,
där r är avståndet mellan partikelcentra; C är; B är en konstant som kännetecknar de frånstötande krafternas energi.
Det är helt uppenbart att på relativt avlägsna avstånd bör attraktionskrafter råda och på nära avstånd frånstötande krafter. På vissa avstånd måste dessa krafter också vara lika, vilket kommer att motsvara den minsta fria energin. Men det är viktigt att notera att under adsorption verkar dispersionskrafter samtidigt mellan varje opolär partikel.
Eftersom interaktionsenergin hos partiklar snabbt kan minska med avståndet, räcker det att utföra summering på de närmaste adsorbentatomerna för att bestämma potentialen för adsorptionskrafter. Det är viktigt att, vid adsorption av komplexa opolära molekyler, den potentiella energin ungefärligen kan beräknas som summan av alla potentiella adsorptionsenergier för molekylens enheter.
Om adsorbenten består av joner, kan verkan av redan kända dispersionskrafter kompletteras med verkan av induktionskrafter för attraktion av dipoler, som induceras i adsorbentens molekyler av ett elektriskt fält, som i sin tur skapas av jonerna i det adsorberande gittret.
Med en sådan växelverkan kan andelen induktiva krafter i adsorptionsväxelverkan vara proportionell mot den adsorberande molekylens polariserbarhet och kvadraten på fältstyrkan på denna adsorberande yta.
Om å andra sidan polära adsorbentmolekyler adsorberas på en polär adsorbent, så polariserar dipolerna i detta fall adsorbentens atomer, d.v.s. som om de inducerar elektriska moment i dem. På grund av denna påverkan läggs den induktiva interaktionen till den dispersions.
Den induktiva växelverkan i sig är vanligtvis liten och kan, beroende på den adsorptiva molekylens dipol och adsorbentens polariserbarhet, nå stora värden. I händelse av att molekyler adsorberas på en adsorbent som har joner eller dipoler på ytan, en s.k. växelverkan mellan joner eller dipoler hos adsorbenten med det elektrostatiska fältet hos själva adsorbenten.
I detta fall kan de adsorberande molekylerna till och med orientera sig i fältet för adsorbenten, och orienterande Coulomb-interaktion inträffar. Det händer vanligtvis att energierna för de induktiva och orienterande interaktionerna är mindre än energin för dispersionsinteraktionen, och därför antas det att energin för intermolekylär attraktion bestäms av energin för dispersionsattraktion.
Dessutom kan bildandet av en vätebindning fungera som en orsak till adsorption. En bindning av denna typ kan uppstå under adsorption på adsorbenter som innehåller hydroxylgrupper av molekyler som vatten, alkoholer, ammoniak och aminer på ytan. När en vätebindning bildas kan interaktionsenergin mellan adsorbenten och adsorbenten vara ganska stor, och värmen som frigörs under sådan adsorption är mycket större än adsorptionsvärmen för ämnen som liknar form och storlek på molekyler, men bildar inte en vätebindning.
Det är viktigt att notera att genom att känna till den termodynamiska beskrivningen av ytskiktet vid "adsorbent - adsorbent" gränsen, dess struktur, arten av olika typer av krafter, dynamiken i processen, kan man gå vidare till studiet av mer komplexa adsorptionsprocesser.
35. Adsorption som spontan koncentration på fasgränsytan av ämnen som minskar gränsytspänningen
Ytaktiva ämnen är indelade i två stora grupper: aktiv och inaktivämnen.
Ytaktiva ämnen kan ansamlas i ytskiktet och i detta fall uppstår positiv adsorption. G > 0.
Sådana typer av ämnen måste ha en ytspänning som i sin tur måste vara mindre än lösningsmedlets ytspänning, annars skulle ansamlingen av ämnet i ytskiktet vara ogynnsam och måste ha en relativt låg löslighet. Med tillräckligt god löslighet tenderar ytaktiva molekyler att lämna ytan djupt ner i lösningen. Därför kommer ytaktiva ämnen företrädesvis att tryckas ut ur huvuddelen av vätskan till ytan.
Men med ackumuleringen av ämnen vid lösningens gräns i molekylerna av dessa ämnen, som interagerar svagt med varandra, kommer den intermolekylära interaktionen i ytskiktet att minska, och ytspänningen kommer att falla.
Ytaktiva ämnen i förhållande till vattenskiktet finns många typer av organiska föreningar, fettsyror med en tillräckligt stor kolväteradikal, salter av dessa syror (tvålar), sulfonsyror och deras salter, samt olika typer av alkoholer och aminer. En karakteristisk egenskap hos de flesta molekyler är deras amfifilitet: molekylen består av två delar av en polär grupp och en opolär kolväteradikal. Att ha ett signifikant dipolmoment och en väl hydratiserande polär grupp kan bestämma det ytaktiva medlets affinitet för den vattenhaltiga miljön. Men kolväteradikalen är orsaken som sänker lösligheten av dessa föreningar.
Inaktiva ytaktiva ämnen- dessa typer av ämnen, tenderar att lämna ytan av vätskan i sin volym, som ett resultat, den så kallade. negativ adsorption G < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Ytanaktiva ämnen i förhållande till vatten finns många oorganiska elektrolyter: syror, alkalier, salter. Molekyler av ytinaktiva ämnen har ingen hydrofob del och kan sönderdelas i vatten till starkt hydratiserande joner.
Exempel ytinaktiva ämnen är också några organiska föreningar där den opolära delen av molekylen är frånvarande eller mycket liten. Dessa ämnen inkluderar myrsyra, aminoättiksyror.
I icke-vattenhaltiga lösningsmedel kan oorganiska elektrolyter också öka ytspänningen, och detta beror på lösningsmedlet.
till exempel När natriumjodid införs i metanol ökar ytspänningen kraftigt, för etanol är ytspänningen ungefär 2 gånger högre. Ämnes ytaktivitet kan bero inte bara på ämnets natur utan också på lösningsmedlets egenskaper. Om något lösningsmedel har en hög ytspänning, kan detta lösta ämne uppvisa betydande ytaktivitet.
36. Adsorptionsteorier
Låt oss överväga de vanligaste adsorptionsteorierna som beskriver enskilda typer av adsorption på gränssnittet "fast gas" eller "fast lösning".
Teorin om monomolekylär adsorption av I. Langmuir.
1. Adsorptionen är lokaliserad och orsakas av krafter nära kemiska.
2. Adsorption sker endast på aktiva centra - utsprång eller fördjupningar på ytan av adsorbenten, kännetecknad av närvaron av fria valenser. Aktiva centra anses vara oberoende och identiska.
3. Varje aktivt centrum kan interagera med endast en adsorbatmolekyl; endast ett lager av adsorberade molekyler kan bildas på ytan.
4. Adsorptionsprocessen är reversibel och i jämvikt; den adsorberade molekylen hålls kvar av det aktiva centret under en tid, varefter den desorberas; Efter en tid etableras en dynamisk jämvikt.
Maximalt möjliga adsorptionsvärde G o uppnås under förutsättning att alla aktiva centra är upptagna av adsorbatmolekyler. Monomolekylär adsorptionsisoterm ekvation som relaterar adsorptionsvärdet G med adsorbatkoncentration Med, ser ut som:
var b- en konstant för ett givet par av "adsorbent - adsorbat"-värde (förhållandet mellan hastighetskonstanterna för desorption och adsorption), numeriskt lika med koncentrationen av adsorbatet, vid vilken hälften av de aktiva centran är upptagna.

Grafen för Langmuir-adsorptionsisotermen visas i figur 2. Konstanten b vi definierar grafiskt genom att rita en tangent till adsorptionsisotermen vid punkten Med= 0. När man beskriver processen för adsorption av gaser i ekvationen kan koncentrationen ersättas med ett proportionellt värde på partialtrycket. Teori om monomolekylär adsorption I. Langmuir tillämplig för att beskriva processerna för adsorption av gaser och lösta ämnen vid låga tryck (koncentrationer) av adsorbatet.
Polanyis teori om polymolekylär adsorption beskriver s-formade adsorptionsisotermer, vars form indikerar möjlig interaktion mellan adsorberade molekyler och adsorbatet.
1. Adsorption orsakas av fysiska krafter.
2. Adsorbentens yta är homogen, det finns inga aktiva centra; adsorptionskrafter bildar ett kontinuerligt kraftfält nära ytan av adsorbenten.
3. Adsorptionskrafter verkar på ett avstånd som är större än adsorbatmolekylens storlek, d.v.s. det finns en viss adsorptionsvolym nära ytan av adsorbenten, som fylls med adsorbatmolekyler under adsorptionen.
4. Attraktionen av en adsorbatmolekyl av adsorbentytan beror inte på närvaron av andra molekyler i adsorptionsvolymen, vilket resulterar i att polymolekylär adsorption är möjlig.
5. Adsorptionskrafter beror inte på temperaturen, och därför ändras inte adsorptionsvolymen med en temperaturförändring.
Freundlichs ekvation. Adsorbentens yta är inhomogen, interaktion sker mellan adsorberade partiklar, aktiva centra är inte helt oberoende av varandra. G. Freindlich föreslog att antalet mol adsorberad gas eller löst ämne per massenhet av adsorbenten (den så kallade specifika adsorptionen X/m), bör vara proportionell mot jämviktstrycket (för gas) eller jämviktskoncentrationen (för ämnen adsorberade från lösning) av adsorbenten upphöjd till en viss styrka, som alltid är mindre än en:
x / m = aP n x / m = aC n.
exponenter n och proportionalitetsfaktor a bestäms experimentellt.
37. Termodynamik för adsorptionsprocessen. Gibbs adsorptionsekvation
För att studera fenomenet med adsorption vid gränsen "lösning - gas", är det nödvändigt att fastställa ett förhållande mellan överskottet av det adsorberade ämnet i skiktet på ytan ( G), koncentration av ytaktiva ämnen i lösning ( med) och ytspänning ( σ ) vid fasgränsen "lösning-gas". Det är mer ändamålsenligt att betrakta fenomenen ur termodynamisk synvinkel och relatera adsorptionen av ett löst ämne till en förändring i ytans fria energi eller dess ytspänning. Denna koppling skapades W. Gibbs i 1876, som namngavs "Gibbs adsorptionsekvation":
G = – med / RT x dσ/dc.
Du kan fortfarande föreställa dig Gibbs ekvation, baserad på termodynamik, med användning av isobarisk-isotermisk potential G kemiska potentialer μ 1 och μ 2 , och även använda n 1 och n 2 antalet mol av komponenterna. Efter att ha analyserat det med hänsyn till entropin S, volym V och tryck P, kan vi skriva följande ekvation:
dG=– SDT+VdP+σds+ μ 1 d n 1 + μ 2 dagar 2.
Vi likställer det med noll, och med hänsyn till konstant temperatur och tryck, förenklas det till en ekvation av formen:
sd σ + n 1 d μ 1 + n2d μ 1 = 0.
Med hänsyn till det faktum att för utspädda lösningar uttrycks den kemiska potentialen för den andra komponenten enligt följande:
μ 2 = μ 2 0 +RT ln c,
och givet att temperaturen är konstant
dμ 2 =rtdnc,
ersätta denna ekvation med
![]()
vi får den önskade Gibbs adsorptionsekvation. Utifrån ekvationen kan man se att om ytspänningen σ ökar med koncentrationen med, då är koncentrationen av det lösta ämnet på ytskiktet mindre än i lösningens volym (den så kallade negativa adsorptionen), och om ytspänningen σ minskar med ökande koncentration med, då är koncentrationen i skiktet större än i volymen (positiv adsorption), och slutligen, om σ beror inte på med, då är koncentrationen av ämnet i skiktet på ytan och i volymen densamma. Gibbs ekvation härleddes med hjälp av termodynamik. Det är svårt att verifiera denna ekvation i praktiken, vilket beror på komplexiteten i att bestämma koncentrationen av ett löst ämne i en skiktad yta. Erfaren B. McBen fann att ett mycket tunt lager vätska skars av från lösningens yta med hjälp av anordningen. Ytterligare bestämning av alla parametrar i Gibbs ekvation visade att de experimentellt hittade värdena för adsorption sammanföll med värdena som beräknades med hjälp av Gibbs ekvation inom det experimentella felet. På grund av homogeniteten och jämnheten hos ytan av någon vätska, när man studerar adsorption på dess yta, är de vanliga idéerna om aktiva centra helt otillämpliga. Vid den kritiska temperaturen försvinner skillnaden mellan de intilliggande faserna, och ytspänningen blir som regel lika med noll. Adsorptionen av gaser och ångor har en så stor praktisk tillämpning att i litteraturen, särskilt i den tekniska, kan du hitta detta koncept, som endast används i förhållande till processer på ytan av fasta ämnen.
Detta koncept, såväl som de mest allmänna adsorptionsmönstren, som den övervägda Gibbs-ekvationen, är tillämplig på alla fasgränser. Med hjälp av Gibbs ekvation och alla bestämmelser som härrör från den, efter att ha bestämt värdet på Г, är det möjligt att konstruera en adsorptionsisoterm.
38. Egenskaper för adsorption på mikroporösa material. Polans potentiella teori. Adsorptionspotential
Glades teoriöverväger icke-lokaliserad fysisk adsorption, som direkt beror på van der Waals-krafter mellan adsorbenten och adsorbatet (detta kan betraktas som den första positionen). Den andra positionen i denna teori är konceptet av kraft- (eller potentiellt) fält för adsorbenten, som sträcker sig över ett avsevärt avstånd från ytan; adsorptionsskiktet som uppträder i detta fält är polymolekylärt. Om vi betraktar adsorptionen av gaser, minskar tätheten av detta lager längs en viss normal från ytan. Om vi överväger ångadsorption, bildas ett vätskeskikt av en viss tjocklek på ytan. Fältet i Polanyis teori betraktas som en serie ekvipotentiella ytor, varje yta motsvarar ett visst värde av potentialen ε , och varje efterföljande yta kommer att vara mindre än den föregående. Varje sådan yta i rymden skär ut lager av en viss volym, betecknad som v i. Uppgiften för Polanyis teori är att hitta övergången från de vanliga koordinaterna för isotermen ( x, sid) till fältparametrar ε i och v i, med ytterligare etablering av sambandet mellan dessa huvudparametrar. Den första delen av problemet, som fastställdes av Polyani, är ganska komplicerad och kan i många fall inte ha säkra lösningar, men för fallet med ångadsorption löses denna del av problemet i den första approximationen mycket enkelt. För ett vätskeadsorptionsskikt kommer den fyllda delen av volymen att vara lika med:
v i \u003d x (M/d),
var där ämnets densitet i flytande tillstånd.
I sin teori introducerar M. Polyany en annan bestämmelse om frånvaron av den sk. fält screening i processen för adsorption, värdet ε i denna teori om rymden är ett konstant värde (något liknande en gravitationspotential) oavsett om vissa adsorbatmolekyler existerar mellan en given punkt och en fast yta, eller om allt utrymme är fritt. Polyani introducerar konceptet adsorptionspotential ε , vilket är det isotermiska arbetet med att komprimera ångan när den överförs från jämviktstrycket R i bulkfasen långt från ytan till området för ytskiktet med mättat ångtryck p 0 då kommer uttrycket för att bestämma potentialen se ut så här:
ε = RT ln R 0 / R.
Med hjälp av en sådan ekvation kan man gå från koordinaterna x, p till koordinaterna ε och v och få en kurva, som kallas "karakteristisk". Polanyi upptäckte i sina experiment att sådana kurvor, konstruerade från experimentella data från de erhållna isotermerna, har följande egenskap: de är invarianta med avseende på T, eller, med andra ord, alla kurvor av denna typ kan ligga på en kurva ε −ε .
M. Polyany accepterade denna position som ett postulat, dvs.:
Denna egenskap hos Polyani är av stor praktisk betydelse, den kan konstruera en familj av isotermer från en experimentell adsorptionsisoterm.
Polanyis teori ger inget analytiskt uttryck för isotermen eller funktionen av potential kontra volym, men låter en beräkna koordinaten för en given temperatur om minst en isoterm är känd. Detta resultat är mycket viktigt för tekniska beräkningar, eftersom för liknande gaser på samma adsorbent kan adsorptionskurvorna visa sig ligga nära varandra och kan i många fall läggas över varandra.
39. Karakteristisk kurva för adsorption. Temperaturinvarians och affinitet för karakteristiska kurvor
Kraftfältet som uppträder vid ytan av adsorbenten kan i många avseenden likna gravitationsfältet. I adsorptionsfältet kan potentiella ytor representeras, d.v.s. ytor för vilka samma adsorptionspotential är karakteristisk. Under begreppet adsorptionspotential θ ska inte förstås som något annat än det arbete som utförs mot adsorptionskrafterna när man flyttar 1 mol av adsorbatet från en viss punkt i fältet till en viss gasfas. Den maximala adsorptionspotentialen kommer att finnas vid gränsen "adsorbent - adsorptionsvolym". Men vid gränsen "volym - gasfas" (det är här verkan av adsorptionskrafter slutar) måste adsorptionspotentialen vara lika med noll. Förändringen i adsorptionspotentialen med en förändring av adsorptionsvolymen kan representeras i form av kurvor. Detta gjordes först av M. Polyani. Sådana typer av kurvor beror inte på temperatur och kan vara karakteristiska för varje specifik adsorbent; sådana typer av kurvor brukar kallas karakteristiska adsorptionskurvor. Teorin om polymolekylär adsorption antar att gasens tillståndsekvation gäller mängden adsorption. Följaktligen liknar de isotermer som kännetecknar beroendet av adsorbatets densitet av volymen för olika temperaturer isotermerna för beroendet av trycket på volymen. Vid låga temperaturer kan adsorptionskrafter på ytan göra att ångan kondenserar till en vätska med en viss densitet. Vid lägre temperaturer än den kritiska, under kondensation, kommer hela adsorptionsvolymen att fyllas med vätska. I detta fall kommer adsorptionskurvan att löpa nästan parallellt med abskissaxeln, vilket är relaterat till vätskans låga kompressibilitet. Sedan sjunker adsorptionskurvan vid gränsen "volym - gasfas" kraftigt nedåt, och följaktligen når densiteten hos adsorbatet värdet av en viss densitet av gasfasen. Vid temperaturer högre än kritiskt kan adsorbenten bete sig som en idealgas, och grafen kommer att uttryckas som en isoterm av beroendet för en idealgas, förutsatt att pV = RT. Under sådana förhållanden kommer den adsorberade gasen att ha en maximal densitet vid själva ytan av adsorbenten och ha en minimal densitet i omedelbar närhet av gasfasen. Dessutom är det i detta fall viktigt att notera att densiteten hos adsorbatet i adsorptionsskiktet ingenstans når densiteten hos själva vätskan. Och om temperaturen är mycket nära kritisk, kommer densitetens beroende av volymen att uttryckas av en kurva nära isotermen, vilket beskrivs van der Waals ekvation. I detta scenario kommer en del av det adsorberade ämnet att vara i den adsorberade volymen i flytande tillstånd, och en del av det adsorberade ämnet kommer att vara i ett gasformigt tillstånd. Då kommer kurvan att minska kraftigast i det avsnitt som motsvarar övergången från vätska till gas. Om en karakteristisk kurva konstrueras från den experimentella adsorptionsisotermen för en av adsorptiva, och man känner till motsvarande affinitetskoefficienter för något annat adsorptiv, kan man hitta adsorptionsisotermen och plotta den för en annan adsorptiv. Potentialteorin för adsorption gör det möjligt att beräkna olika adsorptionsisotermer för olika ångor på samma adsorbent, dessutom enligt den karakteristiska kurvan, som erhålls från adsorptionsisotermen för en ånga, eftersom förhållandet mellan adsorptionspotentialen inte beror på på adsorptionsvolymerna.
affinitet(från latin affinis - "relaterad") - affinitetskromatografi. Metoden för rening och separation av proteiner är baserad på deras selektiva interaktion med en ligand kovalent bunden till en inert bärare (affinitetskromatografi). Att mäta ett toxiskt ämnes affinitet för en receptor är i själva verket en experimentell studie av förhållandet mellan mängden av ett ämne som tillsätts till inkubationsmediet och mängden toxiskt-receptorkomplex som bildas som ett resultat av interaktionen.
Adsorption(från latin ad - på, vid och sorbeo - jag absorberar), en förändring (vanligtvis - en ökning) i koncentrationen av ett ämne nära gränsytan ("absorption på ytan"). Orsak adsorption- omättnad av intermolekylära bindningar nära ytan, d.v.s. förekomsten av ett adsorptionskraftfält. En kropp som skapar ett sådant fält kallas adsorbent, ett ämne vars molekyler kan adsorberas kallas adsorbat och ett redan adsorberat ämne kallas adsorbat. Processen omvänd adsorption kallas desorption.
Arten av adsorptionsfältet är annorlunda. Om adsorption är associerad med van der Waals-bindningar, då adsorption kallas fysisk. Om dessa är valensbindningar, dvs. adsorption passerar med bildandet av ytkemiska föreningar, då adsorption kallas en kemikalie kemisorption. Viktiga funktioner kemisorption visar: irreversibilitet, höga termiska effekter (hundratals kJ / mol), aktiverad karaktär. Det finns många mellantyper adsorption mellan fysikaliskt och kemiskt adsorption. Till exempel, adsorption orsakas av bildandet av vätebindningar. Det finns också olika typer av fysiska adsorption. Den vanligaste förekomsten av dispersionsintermolekylära attraktionskrafter, på grund av det faktum att de är ungefär konstanta för adsorbenter med en yta av någon kemisk natur (ospecifik adsorption). Fysisk adsorption kan orsakas av elektrostatiska krafter (interaktion mellan joner, dipoler eller kvadrupoler); vart i adsorption bestäms av den kemiska naturen hos molekylerna i adsorptiven (den så kallade specifika adsorption). Gränssnittsgeometrin spelar också en viktig roll. om ytan är platt så är den det adsorptionöppen yta, vid lätt eller starkt krökt yta - ca adsorption i adsorbentens porer.
I teorin adsorption Skilj mellan statik (adsorbent-adsorbatsystemet är i termodynamisk jämvikt) och kinetik (det finns ingen jämvikt).
Adsorptionsstatik
Termodynamik för adsorption
.Grunderna i termodynamiken adsorption skapades av J. Gibbs på 70-talet. 1800-talet Enligt Gibbs, i ett tvåfassystem med jämvikt nära gränssnittet, finns det en viss förändring i de lokala värdena för alla omfattande egenskaper (förutom volymen). Faserna anses dock vara homogena upp till någon geometrisk yta som skiljer dem åt. Därför är värdet av en omfattande egenskap för systemet som helhet inte lika med summan av värdena för denna egenskap i homogena faser och . Skillnaden tillskrivs den tvådimensionella ytfasen som är associerad med den separerande ytan. Därför att ytfasen har alltså ingen tjocklek V0=+ och =-, där V- volym.
De presenterade representationerna tillåter oss att reducera den fundamentala termodynamiska ekvationen till formen:
där G är Gibbs fria energi, S är entropin, är gränsytspänningen, s är gränsytan och n i- motsvarande kemiska potential och antalet mol i-den komponenten. Indexet anger värdet på respektive fastighet i ytfasen. Legendre-transformationen låter en modifiera ekvation (1) för isotermiska förhållanden:
Kvantiteten kallas Gibbs adsorption och betecknas med symbolen G, (uttryckt i mol/cm 2). För ett tvåkomponentsystem:
Placeringen av delningsytan kan väljas godtyckligt. I synnerhet kan valet av denna bestämmelse uppfylla villkoret Г 1 =0. En sådan yta kallas ekvimolekylär. För det introduceras beteckningen Г 2 = Г 2 (1). Detta innebär Gibbs huvudsakliga adsorptionsekvation:
Om adsorbenten är helt olöslig i en av de två faserna, =const, och övergången från ekvation (2) till ekvation (3) kräver inte villkoret Г 1 =0. Alltså Gibbs adsorptionär överskottet av denna komponent i ett riktigt tvåfassystem jämfört med ett sådant system där båda faserna skulle vara strikt homogena upp till separeringsytan. Förutom Gibbs överskottsmängder adsorption, spelar en viktig roll i hennes teori adsorption, förstås som hela innehållet i komponenten i i rymden W, som uppvisar adsorptionskrafter. Betecknar hela innehållet genom a och förutsatt att komponenten i helt olösliga i en av bulkfaserna har vi:
där c i-koncentration i-te komponenten i bulkfasen. För små s i:
Adsorption kan förekomma vid vilken gränsyta som helst mellan vilka två faser som helst, i synnerhet vid gränsytan vätska-vätska (vätska-gas, vätska-vätska) eller fast-vätska (fast-gas, fast-vätska). I vätske-vätskesystem kan α mätas som en funktion och experimentellt bestämmas Г 2 (1) med hjälp av ekvation (3). I det andra fallet, för att bestämma G 2 (1) mäts med vilken metod n som helst i 0 , , och koncentrationer av den i:te komponenten i dessa volymer. Härifrån beräknas G i(ett) . Denna metod kallas volumetrisk (eller volumetrisk). Med viktmetoden (gravimetrisk) bestäms mängden direkt i-te komponenten i gränssnittet.
Adsorptionsisoterm
.I ett jämviktsadsorptionssystem är parametrarna som bestämmer jämvikten ett i partiella tryck R(eller med i) och temperatur T. De är relaterade av den så kallade termiska ekvationen:
På adsorption individuell adsorbent ( i=1) denna ekvation har formen:
Tre specialfall av den termiska ekvationen (när T, r eller a- konstanter) spelar en speciell roll i teorin adsorption:
a=- isoterm ekvation adsorption,
T=- isobar ekvation adsorption,
R-- isosterekvation adsorption.
Den specifika typen av funktioner och bestäms av funktionerna i det aktuella systemet. Om en av dem till exempel är känd för något värde T= const, då blir tydligen även de andra två kända. I detta fall är det inte nödvändigt att den analytiska formen av beroenden är känd. De kan ges empiriskt som en uppsättning värden a, r och T.
I teorin adsorption oftast handlar frågan om funktionens form a=(p)r, dvs. om isotermekvationen adsorption. Detta problem är relaterat till termiska effekter som åtföljer adsorption. Vid beräkning av förändringen i värdena för de huvudsakliga termodynamiska funktionerna i fallet med övergången dn mol adsorptivt från bulkfasen till ytfasen i ett jämviktssystem vid p = två fall är möjliga: i det första fallet tas endast hänsyn till omvandlingen av adsorbatet till adsorbatet, eftersom adsorbenten vid adsorption termodynamiskt oförändrad och dess roll är att tjäna som en källa till adsorptionsfältet; i den andra beaktas även förändringen i adsorbenten.
Eftersom systemet är i jämvikt är de kemiska potentialerna för adsorbatet och adsorbatet desamma; adsorbatentropi på grund av en minskning av rörligheten hos molekyler vid adsorption mindre än adsorptivens entropi. Därför, med en inert adsorbent, är entalpin alltid negativ, d.v.s. adsorption exotermisk. Att ta hänsyn till förändringen i entropin hos adsorbenten kan ändra denna slutsats. Till exempel, under polymerernas sorption av ämnen i vilka polymeren sväller, kan entropin hos de senare (på grund av en ökning av rörligheten hos makromolekyler) öka så kraftigt att adsorption blir endotermisk. I det följande endast det exoterma adsorption.
Särskilj integral, differential, isosterisk och medelvärme adsorption. integrerad värme F lika med förlusten av entalpi (vid V= const - konstant intern energi) vid förändring adsorption från en 1 innan en 2(i ett särskilt fall kan det vara en 1 \u003d 0): Q \u003d - (H 2 - H 1). Detta värde refereras vanligtvis till adsorbentens massa och uttrycks i J/kg.
Differentialvärme q(J / mol) är lika med förlusten av entalpi dH när det ändras a på da. Det uttrycks av attityden q = - (dH/da). Det är uppenbart
Isosterisk värme q st tas lika med:
var är skillnaden mellan de molära volymerna av adsorbatet och adsorbatet. Det kan man visa  för en idealisk gasadsorbent:
för en idealisk gasadsorbent:
Innebörden av inledningen qsi genom att dess bestämning inte kräver kalorimetriska data (som t.ex F och q) och det kan beräknas med ekvation (9) från mätresultaten adsorption. Vi introducerar även medelvärmen F(J/mol):
Med tillväxt a parameter F alltid ökande, a q kan minska, öka eller förbli oförändrade. Med tillväxt a med en ojämn yta adsorption förekommer i mindre och mindre aktiva områden, vilket leder till en minskning q. Men i det här fallet minskar de genomsnittliga avstånden mellan de adsorberade molekylerna, vilket resulterar i att attraktionskrafterna mellan dem ökar, och qökar. Förhållandet mellan de två nämnda effekterna avgör beroendets förlopp q=f(a). På mycket stora a frånstötande krafter börjar dominera även i denna region. q minskar alltid med tillväxten a.
För mycket små yttäckningar, isotermekvationen adsorption har formen av Henrys ekvation:
där K H är Henrys koefficient. Faktiskt för mycket små a Adsorptionsskiktet liknar en tvådimensionell idealgas, så dess tillståndsekvation är: rt, där är det tvådimensionella trycket, är det område som upptas av en mol av ämnet. Om vi tar hänsyn till att =- och använder ekvation (3) får vi alltså ekvation (12). Henrys ekvation kräver det q var konstant. För stora fyllningar upphör denna ekvation att hålla. Därför föreslog G. Freindlich (1906) att beskriva isotermer adsorption följande empiriska ekvation (Freundlich-ekvationen):
var k och n- konstanter. Denna ekvation används ofta som en interpolationsformel, även om den är för liten R går inte in i ekvation (12), och i stort R leder till en obegränsad ökning, vilket är oförenligt med erfarenheten a.
Rigorös isotermteori adsorption skapades av I. Langmuir (1914-18). Teorin bygger på följande. modell: 1) ytan av adsorbenten är en uppsättning energiskt identiska aktiva centra på vilka adsorbatmolekyler adsorberas (lokaliseras); 2) endast en molekyl är adsorberad på ett centrum; på adsorption endast en adsorption bildas. skikt (monoskikt); 3) adsorption på detta centrum påverkar inte adsorption på andra centra, dvs interaktion. adsorberade molekyler kan försummas.
Langmuir modell kallas. lokaliserad monomolekylär adsorption på en enhetlig yta. isoterm ekvation adsorption kanske motsvarar den här modellen. fås med hjälp av metoder (molekylär-kinetisk, termodynamisk, statistisk-termodynamisk). Så, adsorption jämvikt kan uttryckas på följande sätt. schema:
Molekylfri. Adsorption i gas + adsorption. komplex fascenter (upptagen centrum)
Koncentrationen av molekyler i gasen är proportionell mot p, koncentrationen av fria. centrum-värde ( a t - a), var och t - totalt antal centra, a-antal ockuperade centra, adsorptionskoncentration. komplex-värde adsorption Därför är jämviktskonstanten: K p \u003d p (a t - a)/ adsorption Härifrån får vi Langmuirs ekvation:
var b-t. kallad adsorption koefficient lika med Kp -1. I området med mycket låga tryck bp" 1 och a = (a m b)p, som motsvarar Henrys ekvation, i vilken KH= a m b. I området med mycket höga tryck br 1 och aa t; vart i adsorption inte längre beroende av tryck. Jämviktskonstant b-1är relaterat till standardvärdet för reaktionens isobariska potential:
Langmuir-modellen kräver att diff. värme och entropi adsorption berodde inte på graden av ytfyllnad.
ekvation (14) är ett strikt uttryck som motsvarar Langmuir-modellen, men det är sällan motiverat i praktiken, eftersom själva modellen är idealiserad adsorption Läran om adsorption från 20-talet 1900-talet i medel. grad byggdes på grundval av att försvaga eller eliminera ett eller annat Langmuir-antagande adsorption
Langmuir har redan föreslagit ett sätt att beskriva adsorption på en inhomogen yta (d.v.s. under antagandet att inte alla centra är lika). Genom att kombinera identiska centra till grupper och anta att ekvation (14) gäller för varje grupp, kan vi anta att adsorptionöver hela ytan uttrycks av summan av termerna i ekvation (14):
Förutsatt att antalet adsorption centra kan vara beskrivs av en kontinuerlig funktion av fördelningen av värdena av gratis. energi, Ya.B. Zel'dovich erhöll från formeln (16) för exponentialfunktionen en ekvation av typen (13).
adsorption på inhomogena ytor - ett stort teorikapitel adsorption Hennes huvudsakliga uppgiftslösning av integralekvationen:
var f(sid) - så kallade. empirisk isoterm adsorption, -det eller en annan f-tion av fördelningen av antalet centra på värdena av gratis. energi,( b, p)- lokal isoterm adsorption, som vanligtvis tas som Langmuir-isotermen adsorption
Många försök har gjorts för att förkasta Langmuirs andra antagande. adsorption På denna väg, teorin om polymolekylär adsorption, föreslagit av S. Brunauer, P. Emmet och E. Teller (BET-teori). Teorin förutsätter att vid en temperatur under den kritiska temperaturen, adsorberas varje molekyl i det första lagret (adsorptionsvärmen q i,), är centrum för molekylerna som bildar det andra lagret, och så vidare. Det antas att värmen adsorption i alla lager, utom det första, är lika med kondensationsvärmen. Denna modell leder till ekvationen:
var c = exp[(q 1 -)/RT]. ekvation (18) i koordinater a, p/p s motsvarar en S-kurva. I koordinater p/p s ,
isoterm adsorption enligt ekvation (18) ska vara linjär. Lutningen för denna räta linje (vanligtvis i intervallet 0,05 p/p s 0,30) och segmentet avskuret av den på y-axeln ger värdena resp. ett t och med. Den utbredda användningen av BET-teorin beror på det faktum att dess författare faktiskt överväger adsorption icke-lokaliserad, identifiera konstanten ett t inte med antalet diskreta adsorbenter. centra, men med antalet adsorbatmolekyler i det första lagret vid närmaste packning (kl R= ps). Därför, genom att introducera idén om området som ockuperas av en molekyl i detta lager, accepterar vi:
var s- adsorbatyta adsorption Som regel mäts isotermen för detta adsorption kväve och ta det för dess molekyl = 0,162 nm 2. En vanligt utförd liknande beräkning s enligt Langmuir-modellen är inte korrekt, eftersom denna metod gäller uppenbarligen bara för icke-lokaliserade adsorption
i teorin om polymolekylär adsorption ett stort bidrag gjordes av J. de Boer, som experimentellt visade att beroendet av det genomsnittliga antalet lager (fler än det första) på alla ytor nära i kemiska termer. naturen, från p/p s uttrycks av en universell kurva (den så kallade t-kurvan). Det gör det också möjligt att uppskatta ytareor av adsorbenter.
Försök gjordes att i Langmuir-modellen även ta hänsyn till interaktionen. mellan adsorbenter. molekyler. Så, T. Hill och J. de Boer, som tror att ekvationen för adsorptionstillstånd. lager är en tvådimensionell analog till van der Waals ekvation, vi har erhållit följande. isoterm ekvation adsorption:
där= a/a t, a och b konstanter för van der Waals ekvation adsorption R. Fowler och E. Guggenheim, med hänsyn till interaktionen. adsorberare molekyler, härledde ekvationen:
där är en konstant associerad med den parvisa interaktionen av molekyler.
Det finns en annan mekanism som leder till ytterligare adsorption adsorbenter under deras kritiska. temperatur på porösa adsorbenter vid relativt höga värden p/p s. Detta är kapillär kondensation. Om en konkav adsorbatmenisk bildas i en por, börjar kondensation i den kl p/p s Enligt Kelvins ekvation:
var är ytspänningen för adsorbatet, V -hans molar volym, r-radie av krökning av menisken adsorption Kapillär kondensering leder till en kraftig ökning av isotermen adsorption I detta fall observeras den så kallade ofta (men inte alltid). adsorption hysteres, dvs. adsorptionsfel. och desorbter. grenar av isotermen. Som regel beror detta på att formen på menisken vid adsorption och desorption stämmer inte överens.
Kapillärkondensation används för att bestämma adsorbentens porstorlek adsorption Enligt ekvation (22) för varje värde p/p s beräkna krökningsradien för menisken adsorption Från det, med tanke på tjockleken på adsorptionen. lager (t.ex. längs t-kurvan), formen på övergångsområdet från lagret till menisken och beroendet av krökning vid mycket liten r , hitta den linjära storleken (effektiv radie r ef) för porerna fyllda vid en given given p/p s. Volymen av sådana porer bestäms av tillväxten adsorption vid denna punkt på isotermen. Med hjälp av de erhållna uppgifterna byggs en porvolymfördelningskurva längs deras radier. Metoden är tillämpbar vid r ef 1,5 nm. Vanligtvis utförs beräkningen genom desorption. grenar av isotermen, men mer strikt modern. teorin kräver att båda grenarna beaktas för att konstruera kurvan.
Potentiella teori om adsorption och teori om volymfyllning av mikroporer. Modell adsorption, fundamentalt skild från Langmuir, föreslogs 1914 av M. Polyaki. Enligt denna modell finns det potentiell adsorption nära ytan av adsorbenten. kraftfältet minskar med avståndet från ytan. Som ett resultat ökar trycket hos adsorptiven, som är lika med p långt från ytan, nära den och når på ett visst avstånd värdet p s vid vilket adsorptiven kondenserar. Volymen av lagret mellan gränssnittet och geomen. plats för punkter där p = p s , fylld med vätska, som tillskrivs de normala värdena av fysisk. bulkvätskeegenskaper. Reversibel isotermisk arbete e adsorption. krafter, bestämda av ekvationen = RTlnp / p s, anropade. adsorption potential, och hela konceptet är en potentiell teori adsorption För en given volym V adsorption skiktet är potentialberoende på temperatur (på grund av dispersionskrafternas oberoende av temperaturen). Denna temperaturinvarians gör det möjligt att räkna om adsorption från ett försök till ett annat, även om isotermekvationerna adsorption det gick inte att utläsa utifrån den angivna teorin. Polyani-modellen har använts flitigt och framgångsrikt av många. författare, men det innehöll två mycket sårbara bestämmelser: 1) antagandet att den finaste adsorptionen. filmen har normala fysiska värden. egenskaper hos bulkvätskan (detta antagande bekräftades inte av experiment); 2) temperaturinvarians för funktionen =f(V), bakom teorin bekräftades ungefärligen genom experiment endast för mycket finporösa adsorbenter.
Med hjälp av potentialteorin har M.M. Dubinin föreslog och utvecklade teorin om volymetrisk fyllning av mikroporer (TOZM). Det har postulerats att denna teori endast gäller mikroporösa adsorbenter. En egenskap hos sådana adsorbenter, i vilka de linjära dimensionerna av porerna är r1 nm, är att hela volymen av deras porer är "fylld" med adsorbenter. fält. Därför, när adsorption de fylls inte i lager, utan volymetriskt. Värdet i det aktuella fallet är inte adsorption. potential, och upp till tecknet för kemikalien. adsorbatpotential, mätt från kemikalienivån. potential hos en normal vätska vid samma temperatur. Hela uppsättningen av adsorbentporer är indelad i tre klasser: mikroporer ( r 0,6 nm), mesoporer (0,6 nm-20 nm) och makroporer ( r 20 nm). adsorption i mikroporer förekommer enligt TOZM-schemat, dvs. volymetriskt, i mesoporer - enligt mekanismen för lager-för-lager fyllning, avslutad med kapillär kondensation. Makroporer under adsorption. jämvikt spelar ingen roll.
Introduktion av konceptet f-tsii-fördelning av porvolymer på värdena av kemikalier. adsorbatpotential i dem, M.M. Dubinin och L. V. Radushkevich erhöll ekvationen för TOZM-adsorptionsisotermen, som vanligtvis skrivs i det följande. form:

var n, E och en 0 -parametrar ( a 0 = a på p = ps). Temperaturberoende a 0:
där= -(da O/dT); en 0 0 = en 0 vid T \u003d T 0. alternativ P och E praktiskt taget oberoende av temperatur. I de flesta fallen P= 2. Endast för fall där de initiala heaten adsorption väldigt stor n > 2. Att räkna om isotermer adsorption från ett adsorptivt till ett annat, antas ungefär att Ei/E2Pi/P= och att a 01 /a 02 V 1 /V 2 , där P i- fallskärm, Vi- molar volym av adsorbent adsorption
Varje mikroporös adsorbent kännetecknas enligt TOZM av två parametrar: W- mikroporvolym ( W 0 = = a 0 V 0) och Eo-karakteristik. energi; W0 och E0 avser standardadsorbenten, vanligtvis bensen.
Använda föreställningen att det finns porer av olika storlekar i en riktig adsorbent, och introducera fördelningen av värden E s varians lika med F. Stekli föreslog en generalisering av ekvation (23), kallad Dubinin-Stöckli-ekvationen:

var B0- konstant förknippas med E i ekvation (23), och y= ![]() . Därför att i adsorptionen tekniken för naib. det var mikroporösa adsorbenter (aktiva kol, zeoliter, finporösa xerogeler) som fick stor spridning, TOZM används inte bara inom fysikaliska och kemiska. forskning, men också i tekniska beräkningar.
. Därför att i adsorptionen tekniken för naib. det var mikroporösa adsorbenter (aktiva kol, zeoliter, finporösa xerogeler) som fick stor spridning, TOZM används inte bara inom fysikaliska och kemiska. forskning, men också i tekniska beräkningar.
Adsorption av gas- och vätskeblandningar. I praktiken handlar de alltid inte om en enskild adsorbent, utan med en blandning av gaser eller flytande lösningar. Därför krävs en generalisering av teorin adsorption för fallet med ett adsorptionsmedel med flera komponenter adsorption I princip kan man utgå från vilken modell som helst adsorption och utvidga det till detta fall. På adsorption gasblandning uppnås detta inte bara genom en större komplikation av ekvationerna, utan också genom att införa tillägg till dem. empirisk parametrar associerade med eller med interaktionen. heterogena molekyler eller, mer allmänt, med inverkan av något in-in på koefficienten. andras aktiviteter. Endast Langmuir-modellen tillåter en att erhålla isotermekvationen adsorption blandningar utan parametrar som inte ingår i ekvationerna för adsorption individuella in-in. För att göra detta är det tillräckligt att ta hänsyn till att under adsorptionen av den kth komponenten från en blandning i komponenter som ingår i adsorptionen. centra kan vara upptas av andra molekyler. Så:
När adsorption flytande lösningar, oavsett deras koncentration, är hela ytan av adsorbenten fylld med adsorption Vari adsorption molekyler av den k-te komponenten åtföljs av förskjutningen av ett visst antal molekyler av de återstående komponenterna, dvs. adsorptionär konkurrenskraftig.
Skilj mellan molekylär och jonisk adsorption lösningar. Den första inträffar när adsorption lösningar av icke-elektrolyter, den andra lösningen av elektrolyter. Molekyl adsorption uttrycks som regel som redundanta värden. Konkurrenskraftig natur adsorption orsakar värdet a m.b. både positiva och negativa. uttrycka adsorption i-av den komponenten som en f-tion av dess molfraktion i lösning x i-, vi har den där G i= O vid x i= 0 och x i= 1 (en möjlig förändring av ämnets volym i adsorptionsskiktet försummas). Därför är isotermen adsorption har en eller flera ytterligheter.
isoterm ekvation adsorption binära lösningar av icke-elektrolyter, tillförlitligt underbyggda termodynamiskt, har formen:

där indexet s indikerar adsorption. fas, - ( dn s 2 /dn s 1) visar hur många mol av den andra komponenten som förskjuts av en mol av den första, skillnaden mellan termerna (standarddelar) av kemikalien. potential som endast beror på temperaturen.
Main problemet med att använda denna och ett antal andra isotermekvationer adsorption-bestämning av koefficientens beroende. aktiviteten hos komponenterna i adsorptionen. lager från dess sammansättning adsorption Den viktigaste frågan i ansökan adsorption för separation eller rening av ämnen - val av en selektiv adsorbent i förhållande till denna komponentlösning adsorption
Jonisk adsorption, är som regel inte likvärdig adsorption Preim adsorberas på ytan från elektrolytlösningen. katjoner eller anjoner. Tack vare det elektriska (Coulomb) krafter på ytan bildas elektriskt dubbelskikt.
Om adsorbenten innehåller joner eller ytfunktion. grupper som kan joniseras i ett givet lösningsmedel, då sker jonbyte mellan adsorbenten och elektrolytlösningen. Adsorbenten i detta fall kallas. jonbytare.
Adsorptionskinetik
adsorption, som alla verkliga processer, inträffar i tiden. Så hela teorin adsorption bör innehålla ett avsnitt om kinetik adsorption elementär handling adsorption utförs nästan omedelbart (med undantag för kemisorption). Alltså tidsberoendena adsorption definieras i huvudsak diffusionsmekanism, dvs tillförsel av adsorptivmedel till platsen adsorption Om en adsorption på den öppna ytan inte är ögonblicklig, en sådan process inträffar i det externa diffusionsområdet; medan diffusionslagarna inte är specifika för adsorption När det gäller porösa adsorbenter, förutom ext. diffusion, en viktig roll börjar spela vnutr. diffusion, dvs. överföring av adsorbenten i adsorbentens porer i närvaro av en koncentrationsgradient i dem. Mekanismen för sådan överföring kan bero på adsorbatkoncentrationen och porstorlekarna.
Det finns molekylär-, Knudsen- och ytdiffusion (Volmer). Molekylär diffusion utförs om längden är fri. intervallet av molekyler i porerna är mindre än porstorleken, Knudsen-längden är om denna längd överstiger porstorleken. Under ytdiffusion rör sig molekyler över ytan av adsorbenten utan övergång till bulkfasen. Men värdena för koefficienten diffusioner är inte desamma för olika diffusionsmekanismer. I många fall är det inte möjligt att experimentellt fastställa exakt hur diffusion sker, och därför s.k. effektiv koefficient. diffusion, som beskriver processen som helhet.
Main experimentell material om kinetik adsorption betjänar den sk. kinetisk kurva, dvs. f-tion \u003d a / a lika \u003d med) var är relativt adsorption lika med förhållandet mellan det aktuella värdet av adsorption a till a lika med dess värde vid tidpunkten t. Att tolka kinetiken kurva i det enklaste fallet antas det att adsorbentkornet har en helt likformig porös struktur i volym (denna modell kallas kvasihomogen). betyder att. förbättring av den kvasihomogena modellen - uppfattningen att varje korn innehåller regioner med större och finare porer. Diffusion i ett sådant korn beskrivs av två dec. koefficienter.
I fallet med en öppen yta, med Langmuir-modellen, är det lätt att få kinetiken. ekvationen adsorption Hastigheten för närmande till jämvikt är skillnaden i hastigheter adsorption och desorption. Om vi antar, som vanligt inom kinetiken, att processhastigheterna är proportionella mot koncentrationerna av reagerande ämnen, har vi:
där k ads och k dec är respektive hastighetskonstanter. adsorption och desorption. Trycket i gasfasen antas vara konstant. När man integrerar denna ekvation från t= 0 till vilket värde som helst t vi får:
Därför har vi för f:= lika. Så vi har äntligen:
där k = k annonser + k dec.
Temperaturens inverkan på hastigheten adsorption uttrycks med en ekvation som liknar Arrhenius-ekvationen adsorption Med ökande temperatur ökar k ads exponentiellt. Därför att diffusion i adsorbentens porer är förknippad med att övervinna aktivering. barriärer, temperaturberoendena för k ads och k des är inte desamma.
Kunskap om diffusionshastigheter är viktig inte bara för teori adsorption, men också för beräkning av bal. adsorption processer. I det här fallet handlar de vanligtvis inte om enskilda korn av adsorbenten, utan med deras lager. Kinetiken för processen i lagret uttrycks av mycket komplexa beroenden. Vid varje punkt i lagret vid en given tidpunkt, värdet adsorption bestäms inte bara av isotermekvationens form adsorption och lagarna för processens kinetik, men också aero- eller hydrodynamisk. förhållanden för flödet av gas eller vätska runt spannmål. Kinetiken för processen i det adsorberande skiktet, i motsats till kinetiken i ett enda korn, kallas. dynamik adsorption, vars allmänna schema för att lösa problem är som följer: ett system med differentialer sammanställs. ekvationer i partiella derivator, med hänsyn tagen till skiktets egenskaper, isotermen adsorption, diffusionsegenskaper (diffusionskoefficient, typer av massöverföring längs lagret och inuti kornen), aero- och hydrodynamiska. flödesfunktioner adsorption Initial- och randvillkor är fastställda. Lösningen av detta ekvationssystem leder i princip till värdena för kvantiteterna adsorption vid en given tidpunkt vid en given punkt i lagret. Som regel analytisk lösningen kan endast erhållas för de enklaste fallen; därför löses ett sådant problem numeriskt med hjälp av en dator.
I en experimentell studie av dynamiken adsorption en gas- eller vätskeström med specificerade egenskaper leds genom adsorbentskiktet och sammansättningen av den utgående strömmen undersöks som en funktion av tiden. Utseendet på det absorberade ämnet bakom lagret kallas. genombrott, och tiden till genombrott - tiden för skyddsåtgärden. Beroendet av koncentrationen av denna komponent bakom lagret på den anropade tiden. utgångskurva. Dessa kurvor fungerar som huvud experimentell material som gör det möjligt att bedöma dynamikens mönster adsorption
Hårdvarudesign av adsorptionsprocesser
Det finns många tekniker. adsorptionstekniker. processer. Utbredd cyklisk. (periodiska) installationer med fast adsorbentbädd, osn. vars nod är en eller flera. adsorbatorer tillverkade i form av ihåliga kolonner fyllda med granulär adsorbent. En gasström (eller vätskeström) innehållande adsorberade komponenter leds genom adsorbentbädden tills genombrott adsorption Därefter regenereras adsorbenten i adsorbatorn och gasflödet skickas till en annan adsorbator. Adsorbentregenerering innefattar ett antal steg, varav det huvudsakliga är desorption, d.v.s. frigöring av tidigare absorberat material från adsorbenten adsorption Desorption utförs genom uppvärmning, trycksänkning av gasfasen, förskjutning (t.ex. levande ånga) eller en kombination av dessa metoder. Eftersom tider adsorption och regenerering inte sammanfaller, välj ett sådant antal samtidigt arbetande och regenererade adsorbatorer att hela processen pågår kontinuerligt.
Enligt tech. och ekonomiskt överväganden förnyelse inte avslutas adsorption Därför är adsorbentens arbetskapacitet lika med skillnaden mellan det maximalt uppnåbara under givna förhållanden adsorption och mängden adsorbat som finns kvar i adsorbenten efter regenerering. Som ett resultat är isotermerna adsorption som motsvarar processen i adsorbern bör inte vara för brant.
I det beskrivna schemat är två alternativ möjliga: 1) målprodukten adsorberas från gasströmmen nästan fullständigt, och sedan ingår den i desorbatet, varifrån den extraheras på ett eller annat sätt; 2) målprodukten adsorberas sämre än andra komponenter i gasblandningen, och sedan ingår den i den utgående gasströmmen. Enligt det första alternativet fungerar till exempel återvinningsenheter på viskosfabriker, fångar upp från avgaser och återför CS 2 till cykeln. Produktiviteten hos sådana installationer når hundratusentals m 3 renad gas per timme; adsorberande aktivt kol med inte alltför fina mikroporer, dvs. kol, där konstanten E enligt TOZM (se ovan) är 20-25 kJ / mol. Detta värde E 0 motsvarar en inte alltför brant isoterm, vilket ger goda regenereringsförhållanden. Sådana kol kallas återhämtning. Desorption utförs med levande ånga. För att spara energi leds kalla och heta gasflöden genom värmeväxlare.
Det är mycket viktigt att torka gaser och vätskor, såsom petroleumgaser, innan de bearbetas eller naturen. gaser före transport; silikageladsorbenter eller zeoliter. Desorption utförs genom uppvärmning. Eftersom desorptionen av zeolit är förknippad med höga energikostnader, används en kombinerad adsorbent: huvud. fuktmassa absorberas av lätt regenererad silikagel och djup eftertorkning av zeolit.
Under termisk regenerering inkluderar hela cykeln adsorption adsorberande uppvärmning, dess desorption och kylning. Ett stort antal steg bestämmer processens låga intensitet och höga energiintensitet. adsorption Därför är den sk. kortcykelinstallationer, hela cykeln i vilken tar flera. minuter. I dem tillförs gas till adsorbatorn under medelvärdet. tryck, som sedan släpps och desorption sker. Hela processen är nästan isotermisk (avvikelse från isotermalitet orsakas endast av frigöring av värme adsorption och absorption av värme under desorption). Cykelsteg: adsorption, trycksänkning, desorption, tryckstegring. Ett exempel är en anläggning med zeolit för att producera syreberikad luft.
I installationer med ett rörligt skikt av adsorbent (i de så kallade hypersorberarna), sjunker den senare långsamt under påverkan av gravitationen, tas bort från botten. delar av adsorbatorn och går in i den sk. airlift, som är ett vertikalt rör parallellt med adsorption. kolumn. Ett luftflöde rör sig genom detta rör nedifrån och upp, vilket höjer adsorbentkornen till toppen. del av kolumnen. Det bearbetade gasflödet kommer in i den mellersta delen av adsorbatorn och rör sig upp i motström till adsorbenten. Överst i kolonnen finns en kontinuerlig adsorption, längst ner - regenerering av adsorbenten (se även adsorptionsrengöring).
I anläggningar med en fluidiserad ("kokande") adsorbentbädd bringar gasflödet som kommer in i adsorbatorn underifrån adsorbenten i suspension. Detta ökar kraftigt effektiviteten av massöverföring mellan adsorbenten och gasen och minskar varaktigheten adsorption och desorption. Sådana installationer har hög produktivitet. Deras breda spridning hindras av höga krav på päls. hållfastheten hos adsorbentkornen (otillräcklig styrka orsakar betydande förluster av adsorbenten på grund av dess nötning och medryckning från apparaten).
Main krav på adsorbenter: stor adsorbent. kapacitet, dvs. de borde vara spridda kroppar med ett stort slag. yta eller med stor porvolym; chem. ytans beskaffenhet måste ge effektiv adsorption data in-in under dessa förhållanden; chem. och termisk. hållbarhet, regenererbarhet, tillgänglighet. max. aktiva kol, xerogeler av vissa oxider (kiselgeler, aluminiumoxidgeler, etc.), zeoliter har blivit utbredda; från icke-porösa adsorbenter-tech. kol (sot) och högdispergerad SiO 2 (aerosil, "vit sot").
Användningsområden för adsorptionsteknik
Om fenomenet adsorption grundat av många sätt att rengöra luften från skadliga föroreningar (se. gasrening), vatten (se Vattenbehandling), samt sockersirap för sockertillverkning, fruktjuicer och andra vätskor i mat. prom-sti, bortfall av smörjoljor. Att ta bort fukt som en skadlig förorening från gaser och vätskor med hjälp av fasta adsorbenter är en av de viktiga grenarna av adsorption. tekniker (se även gastorkning).
På adsorption. processer är baserade på finseparering av blandningar av ämnen och isolering av vissa komponenter från komplexa blandningar. Exempel är separation av isomerer av alkaner för att erhålla normala kolväten för framställning av ytaktiva ämnen, separation av oljor vid framställning av motorbränslen. För gasblandningar av adsorption. separationsmetoder används för att erhålla luft berikad med syre (upp till nästan ren O 2); i många fall konkurrerar dessa metoder framgångsrikt med destillation (se. luftseparering).
Det snabbt växande användningsområdet för adsorbenter. medicinsk teknik, där den tjänar till att utvinna skadliga ämnen ur blodet (hemosorptionsmetoden), etc. fiziol. vätskor. De höga kraven på sterilitet utgör en mycket svår uppgift att välja lämpliga adsorbenter. Dessa inkluderar speciellt framställda aktivt kol.
Belyst.: Brunauer S., Adsorption av gaser och ångor, trans. från engelska, vol. 1, M., 1948; de Boer Ya, Adsorptionens dynamiska natur, trans. från English, M., 1962; Adsorption and Porosity, red. M.M. Dubinina [et al.], M., 1976; Keliev N.V., Fundamentals of adsorption technology, 2nd ed., M., 1984; Young D.M., Crowell A.D., Physical adsorption of gases, L., 1962. M.M. Dubinin, V.V. Serpinsky.
Välj första bokstaven i artikelns rubrik:
När det gäller växelverkan mellan två atomer:
U är interaktionsenergin;
U = U + U DRAG.
 - Lennard-Jones ekvation
, c, b, m = konst
- Lennard-Jones ekvation
, c, b, m = konst
I fall av interaktion av atomer med en fast yta är det nödvändigt att summera alla interaktioner.
x är avståndet till ytan
r - aktionsradien för attraktionskrafterna
dV - volym
n är antalet ytmolekyler
U ADS. är adsorptionsinteraktionsenergin



Vid adsorption förstärks attraktionen. Och i fallet med interaktion av den opolära-opolära typen är adsorptionen övervägande lokaliserad i urtagningarna.
elektrostatisk interaktion.
Polär adsorbent - opolärt adsorbat
Icke-polär adsorbent - polärt adsorbat
En polär adsorbent är ett polärt adsorbat.
M  adsorbatmolekylen representeras som en dipol och adsorbenten som en ledare, i vilken adsorbatmolekylen inducerar en dipol spegelsymmetriskt med avseende på den givna.
adsorbatmolekylen representeras som en dipol och adsorbenten som en ledare, i vilken adsorbatmolekylen inducerar en dipol spegelsymmetriskt med avseende på den givna.
X - avstånd till mitten
När du interagerar uppstår potentialen:
 ,
,
 är dipolmomentet.
är dipolmomentet.
Potentialen tenderar att ta på maxvärdet, d.v.s. dipoler tenderar att orientera sig vinkelrätt mot ytan.
Eftersom en ökning av temperaturen främjar tillväxten av Brownsk rörelse, leder det till en bromsning av adsorptionsprocessen.
Vid elektrostatisk interaktion är adsorbatet övervägande lokaliserat på utsprång.
Grundläggande adsorptionsekvation.
Vid adsorption omfördelas komponenten, vilket innebär att den kemiska potentialen förändras. Adsorptionsprocessen kan betraktas som övergången av ytenergi till kemisk energi.
Skiktvolym = 0, sedan den generaliserade ekvationen I och II för termodynamikens lag:
T = const; (1) = (2) => 

För ett tvåkomponentsystem:
,
 ,
,
 =>
=>
=>
 - Gibbs adsorptionsekvation
.
- Gibbs adsorptionsekvation
.
För fallet med adsorption av TV. kropp - gas:, 
 ,
,

 - isoterm
- isoterm
 - isobar
- isobar
 - isopykne
- isopykne
 - isoster
- isoster
Isoterm, isopycne, isostere är släkt med varandra.
Därför att adsorptionsfunktion 



Henry isoterm Langmuir isoterm



Termodynamik. Adsorption.
För kondenserad media:
 ,
,
 ,
,
 - integrerad förändring i Gibbs energi
.
- integrerad förändring i Gibbs energi
.

P-tryck över en krökt yta, P S-tryck över en plan yta
 - adsorptionspotential
- adsorptionspotential
Differentiell förändring i entrapi 
 , Г = konst
, Г = konst
- differentiell entropiförändring
- differentiell entalpi för adsorption
 - isosterisk adsorptionsvärme
- isosterisk adsorptionsvärme
 - kondensationsvärme
- kondensationsvärme
 - nettoadsorptionsvärme
- nettoadsorptionsvärme

 ,
,




Qa är det integrerade adsorptionsvärmet, 
Qra är den integrerade nettovärmen för adsorption,

Henrys ekvation
Studiet av adsorption hämmas av ytans inhomogenitet, så de enklaste regelbundenheterna erhålls för homogena ytor.
Låt oss betrakta växelverkan mellan gaser och en fast yta, när en gas övergår från ett jämviktstillstånd i volymen till ett jämviktstillstånd på ytan. Detta fall är analogt med jämvikten mellan gaser i ett gravitationsfält.
 ,
,
 ,
=>
,
=> -Henrys ekvation
-Henrys ekvation
 - Fördelningskoefficient
- Fördelningskoefficient
I adsorptionsprocessen sker en förändring i kemiska potentialer.
För bulkfas: 
För ytgas: 
I ett tillstånd av jämvikt  , dvs.
, dvs.

I Henrys ekvation beror konstanten inte på koncentrationen
Henrys ekvation är giltig i området med låga tryck och koncentrationer. När koncentrationen ökar är två typer av avvikelser från Henrys lag möjliga:
1 - positiva avvikelser, D minskar, A minskar
2 - negativa avvikelser, D - ökar, A - ökar.
Typen av avvikelse bestäms av dominansen av en eller annan typ av adsorbent-adsorbatinteraktion.
Med en stark adhesiv interaktion ökar aktivitetskoefficienterna - en positiv avvikelse. Vid kohesiva interaktioner observeras negativa avvikelser.
monomolekylär adsorption.
Langmuir isoterm.
De enklaste regelbundenheterna erhölls i Henrys teori. Langmuir föreslog en teori enligt vilken adsorption betraktas som en kvasikemisk reaktion. Vart i:
Ytan är energimässigt enhetlig.
Adsorptionen är lokaliserad, varje adsorptionscentrum interagerar med en adsorbatmolekyl.
Adsorbatmolekyler interagerar inte med varandra.
Adsorption är monolager.

 - yta,
- yta,  - adsorbat,
- adsorbat,  - adsorptionskomplex.
- adsorptionskomplex.
 , sedan koncentrationen av adsorptionsställen:
, sedan koncentrationen av adsorptionsställen:  ,
, - begränsa adsorptionen.
- begränsa adsorptionen.
 , sedan reaktionskonstanten:
, sedan reaktionskonstanten: 

 - Langmuirs ekvation.
- Langmuirs ekvation.
Adsorption kontra koncentration
1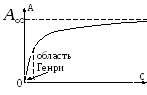 )
)
 ,
,
2) område med höga koncentrationer
 - begränsande adsorption, bildning av ett monomolekylärt skikt
- begränsande adsorption, bildning av ett monomolekylärt skikt
För Gibbs energi: .
g är entropifaktorn.
I fallet med Henry-isotermen karakteriserar Gibbs-energin övergången av adsorbatet från standardtillståndet i bulken till standardtillståndet på ytan. I fallet med Langmuir-isotermen  kännetecknar graden av affinitet för adsorbenten och adsorbatet.
kännetecknar graden av affinitet för adsorbenten och adsorbatet.
 hittat från van't Hoff isobar.
hittat från van't Hoff isobar.

 , då
, då  , därav
, därav  .
.

 - grad av ytfyllnad.
- grad av ytfyllnad.




 - antalet lediga platser,
- antalet lediga platser,  - antalet upptagna platser.
- antalet upptagna platser.
 ,
,

De där. i området med höga koncentrationer är antalet fria ställen omvänt proportionellt mot mängden adsorbat.
Adsorption av en blandning av gaser på en homogen yta.
I detta fall betraktas adsorptionsprocessen som två parallella reaktioner.
 (1)
(1)

 (2)
(2)


Adsorption av en blandning av gaser på en inhomogen yta.
Vid en ohomogen yta bör man inte begränsa sig till medelstora fyllningar.
Som ett resultat av konkurrens är lokalisering av olika adsorbater möjlig på olika typer av platser.
I detta fall förhållandet  .
.
 ,
,
 är mättnadsångtrycket för adsorbatet.
är mättnadsångtrycket för adsorbatet.
 ,
,
 är adsorptionsvärmen.
är adsorptionsvärmen.
"+" - symbatiskt beroende, "-" - antibatiskt beroende, "H" - ingen korrelation.
"+" - adsorption fortskrider enligt samma mekanism. I de mest energimässigt gynnsamma områdena adsorberas en gas med hög affinitet till ytan övervägande.
"-" - adsorption fortskrider genom olika mekanismer och fram till en viss tidpunkt finns ingen konkurrens om ytan.
Monomolekylär adsorption realiseras huvudsakligen under den fysiska adsorptionen av gaser vid låga värden sid, såväl som vid vätske/gas-gränsytan.
Polymolekylär adsorption.
BET teori(Brunauer, Emmet, Teller).
I det fall då bildningen av ett monoskikt är otillräcklig för att kompensera för ytenergin, är adsorption polymolekylär och kan betraktas som ett resultat av påtvingad kondensation under inverkan av ytkrafter.
Grundläggande bestämmelser:
När en adsorbatmolekyl träffar den ockuperade platsen bildas en multipel uppsättning.
När du kommer närmare sid till sid s antalet fria adsorptionsplatser minskar. Inledningsvis ökar antalet platser som upptas av singel, dubbel etc. för att sedan minska. kit.
På sid =sid s adsorption övergår i kondensation.
Det finns inga horisontella interaktioner.
För det första lagret utförs Langmuir-isotermen.
Ytan betraktas som en uppsättning adsorptionsställen. Villkoret för dynamisk jämvikt är giltigt: kondensationshastigheten på fria platser är lika med avdunstningshastigheten från ockuperade.
a är kondensationskoefficienten (fraktionen av molekyler som kondenseras på ytan);
 ,
,
Zm är det maximala antalet lediga platser.
 - frekvens av vibrationer av atomer i riktning vinkelrät mot ytan.
- frekvens av vibrationer av atomer i riktning vinkelrät mot ytan.
För det första lagret är de dynamiska jämviktsförhållandena:



 , då
, då
 - Langmuirs ekvation.
- Langmuirs ekvation.
För det andra lagret kommer att vara sant: 
För det i-te lagret: 
För enkelhetens skull antas det att a och ν är lika för alla lager utom det första. För alla lager utom det första är adsorptionsvärmen konstant. För det sista lagret är adsorptionsvärmen lika med kondensationsvärmen. Som ett resultat, ekvationen
 (*)
(*)
C- konstant,
När det gäller BET-teori, konstanten Med kännetecknar Gibbs energi av ren adsorption. Ekvationen innehåller bara en konstant, och denna ekvation är också mycket viktig för att bestämma adsorbentens specifika yta.
Eftersom värme frigörs som ett resultat av adsorption, utförs bestämningen av specifika ytor vid låga temperaturer.
 ????????????
????????????


Den största bristen i teorin– Försummelse av horisontella interaktioner till förmån för vertikala.
Ekvationen ligger inom intervallet  från 0,05 till 0,3.
från 0,05 till 0,3.
Var  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 - interaktionen av adsorbatet - adsorbat påverkar.
> 0,3 - interaktionen av adsorbatet - adsorbat påverkar.
Redovisning av adsorbat-adsorbat-interaktioner.
Interaktioner uppträder under adsorption på en opolär yta av grenade molekyler eller molekyler. kan bilda föreningar. I detta fall ändras formen på adsorptionsisotermerna.
MEN  sorbenten är inte polär.
sorbenten är inte polär.
Diagram 1 motsvarar svaga interaktioner adsorbat-adsorbat, starkt adsorbat-adsorbent.
Diagram 2 motsvarar en stark adsorbat-adsorbat-interaktion, en stark adsorbat-adsorbent-interaktion.
Diagram 3 motsvarar en stark adsorbat-adsorbat-interaktion, en svag adsorbat-adsorbent-interaktion.
 ,
,

Vid interaktion mellan adsorbatmolekyler är det nödvändigt att ta hänsyn till förändringar i aktivitetskoefficienterna. Och denna ekvation är skriven som:
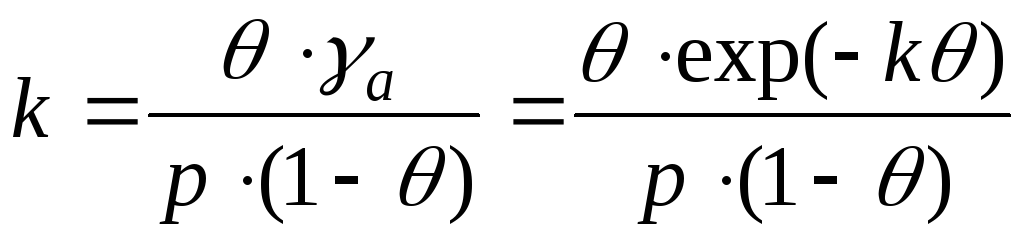 - Frunkins, Fowlers, Guggenheims ekvation.
- Frunkins, Fowlers, Guggenheims ekvation.
kär attraktionskonstanten.
Polans potentiella teori.
Denna teori härleder inte någon typ av adsorptionsisoterm, men gör det möjligt att beräkna isotermer vid en annan temperatur.
Adsorptionär resultatet av attraktionen av adsorbatet till ytan av adsorbenten på grund av adsorptionspotentialens verkan, som inte beror på närvaron av andra molekyler och beror på avståndet mellan ytan och adsorbatmolekylen.
 ,
,
 - adsorptionspotential.
- adsorptionspotential.
Eftersom ytan är inhomogen ersätts avståndet med adsorptionsvolymen  .adsorptionsvolymär volymen innesluten mellan ytan och punkten som motsvarar det givna värdet
.adsorptionsvolymär volymen innesluten mellan ytan och punkten som motsvarar det givna värdet  .
.
Adsorptionspotentialär arbetet med att överföra 1 mol av adsorbatet utanför den givna adsorptionsvolymen till en given punkt av adsorptionsvolymen (eller arbetet med att överföra 1 mol mättad ånga av adsorbatet, som är i jämvikt med det flytande adsorbatet i frånvaro av adsorbenten, in i ångfasen i jämvikt med adsorbenten).

Karakteristisk kurva
 - adsorptionspotential,
- adsorptionspotential, 
För en given adsorbent och olika adsorbater gäller följande: 
För olika typer av adsorbater  ,
,
var  potentialer för adsorptionsisotermer vid relativa tryck
potentialer för adsorptionsisotermer vid relativa tryck  för adsorbat 1 och för adsorbat 2. Detta förhållande är ett konstant värde.
för adsorbat 1 och för adsorbat 2. Detta förhållande är ett konstant värde.
 - affinitetskoefficient
- affinitetskoefficient
Teori om kapillär kondensation.
Förloppet av adsorptionsprocessen beror till stor del på strukturen hos den porösa kroppen.
|
mikroporösa | |
|
Övergångs porös | |
|
Makroporös |
I fallet med mikroporösa sorbenter överlappar fälten av adsorptionskrafter. När det gäller makroporösa sorbenter fungerar porerna som transportkanaler. Kondensationsprocesserna är mest betydande i transienta porösa kroppar. Kapillärkondensering startar vid vissa värden sid och  när en del av ytenergin redan har kompenserats. En nödvändig förutsättning är att ytan ska vara självbärande. Processen beskrivs Thompson-Kelvins ekvation.
när en del av ytenergin redan har kompenserats. En nödvändig förutsättning är att ytan ska vara självbärande. Processen beskrivs Thompson-Kelvins ekvation.

 - för vätning är krökningscentrum i gasfas.
- för vätning är krökningscentrum i gasfas.
Vid kapillärkondensation har adsorptionsisotermen en hysteresform. Den nedre grenen motsvarar adsorptionsprocessen och den övre grenen motsvarar desorptionsprocessen.
Alla typer av porer kan reduceras till tre typer:
|
konisk |
Cylindrisk med en sluten ände |
Cylindrisk med två öppna ändar |
|
Processfyllning utförs från botten av poren. Adsorptionsisotermen och desorptionsisotermen sammanfaller i detta fall, eftersom adsorptionsprocessen börjar med en sfär och desorptionsprocessen också börjar med att vissa sfärer försvinner.
↓ |
Det finns ingen hysteres. Slag framåt och bakåt beskrivs av ekvationen:
|
Det finns ingen botten någonstans, fyllningen av poren kommer att gå längs cylinderns väggar.
cylinder: Isoterm och kommer att ha en hysteresform.
↓ |


 - sfär,
- sfär, ,
,



PÅ  under väta förhållanden sker kondens vid lägre tryck, vilket är energetiskt gynnsamt. Från desorptionsgrenen erhålls porstorleksfördelningskurvor.
under väta förhållanden sker kondens vid lägre tryck, vilket är energetiskt gynnsamt. Från desorptionsgrenen erhålls porstorleksfördelningskurvor.
Differentialkurvans maximum förskjuts åt vänster i förhållande till integralens inflexionspunkt. Den totala volymen av små porer är liten, men har stora ytareor. När porstorleken ökar, ökar deras volym som  , och området som
, och området som  På grund av detta observeras en förskjutning av differentialkurvans maximum.
På grund av detta observeras en förskjutning av differentialkurvans maximum.
Adsorption vid gränssnittet mellan fast och vätska.
När det gäller adsorption vid fast-gas-gränssnittet försummade vi en komponent. I fallet med adsorption vid gränsytan mellan fast och vätska, tränger adsorbatet undan lösningsmedelsmolekyler från ytan av adsorbenten.
 ,
,
Den högra ekvationen är:

 ,
,
N 1, N 2 - molfraktioner av lösningsmedlet och komponenten, N 1 + N 2 \u003d 1, sedan
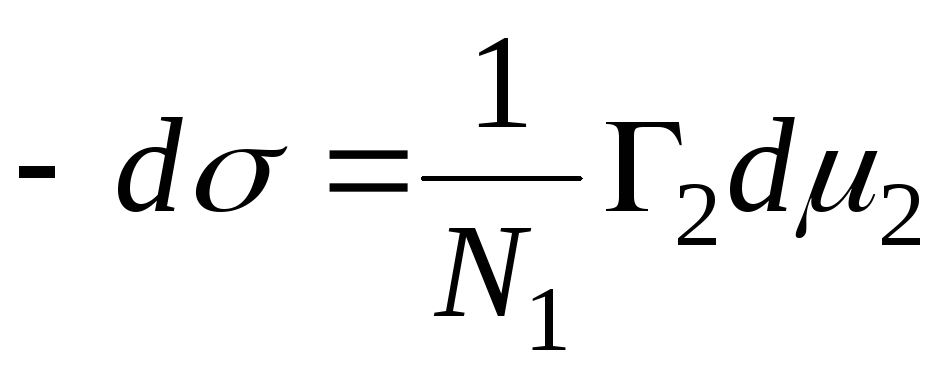 ,
=>
,
=>
 , sedan - adsorptionsekvationen för fasgränsen fast - vätska.
, sedan - adsorptionsekvationen för fasgränsen fast - vätska.
Adsorption (G) > 0 at  <
0
<
0
Om värdena  för komponenten och lösningsmedlet är mycket olika, i detta fall beroendet G från N har ett extremum vid värdet N
~ 0,5.
för komponenten och lösningsmedlet är mycket olika, i detta fall beroendet G från N har ett extremum vid värdet N
~ 0,5.
E  om
om  har liknande värden, i detta fall kan tecknet på adsorption ändras. Missbruk G från N korsar x-axeln
har liknande värden, i detta fall kan tecknet på adsorption ändras. Missbruk G från N korsar x-axeln
Funktion Korsning G(N) med abskissaxeln kallas adsorptionsazeotrop. Detta innebär att de två komponenterna inte kan separeras på denna adsorbent.
Adsorptionsisotermekvation med utbyteskonstant.
Under adsorption vid gränsytan mellan fast och vätska omfördelas komponenterna konstant mellan ytan av adsorbenten och volymen av lösningen.
 - komponenter (- - hänvisar till ytan)
- komponenter (- - hänvisar till ytan)

 ,
,
 ,
, .
.
 ,
,


Adsorption vid vätske-gas-gränsytan
R  Låt oss betrakta förändringen i koncentrationsprofilen när vätske-gasgränsytan korsas. Låt komponent 2 vara flyktig.
Låt oss betrakta förändringen i koncentrationsprofilen när vätske-gasgränsytan korsas. Låt komponent 2 vara flyktig.
Cs är koncentrationen i ytskiktet.
Baserat på definitionen av överskottsadsorption

Om komponenten inte är flyktig, kommer adsorptionsvärdet att skrivas enligt följande:
P  ri
ri 

I ekvationen  materiens natur beskrivs av derivatan
materiens natur beskrivs av derivatan  .
.
Ytspänningsisotermen kan vara av formen 1 eller 2:
1 - ytaktiva ämnen
2 - ytaktiva ämnen
Ytaktivitet g är ämnens förmåga att minska ytspänningen i systemet.

 - tjockleken på ytskiktet
- tjockleken på ytskiktet
C sär koncentrationen av komponenten i ytskiktet
Med– volymkoncentration
För en homolog serie finns en regel:
 - Traubeau Duclos härskar
- Traubeau Duclos härskar
För den homologa serien ser adsorptionsisotermen ut så här:
Vi skriver D istället för A, eftersom adsorptionen är för stor i ytskiktet.
Ytspänningsisoterm:

 är ytspänningen för det rena lösningsmedlet.
är ytspänningen för det rena lösningsmedlet.
 - fundamental adsorptionsekvation;
- fundamental adsorptionsekvation;
 - Langmuirs ekvation.
- Langmuirs ekvation.
Låt oss lösa dem tillsammans:

- Shishkovskys ekvation.
Bär en konstant för den homologa serien.
A- när man flyttar från en homolog till en annan ökar den med 3-3,5 gånger 
![]()
1 - område med låga koncentrationer
![]()
2 - medelkoncentration
3 - monomolekylärt skikt
Ytaktiva ämnen är amfifila molekyler, dvs. inkluderar en polär grupp och en opolär kolväteradikal.
o är den polära delen av molekylen.
| är den opolära delen av molekylen.
I ett polärt lösningsmedel är ytaktiva molekyler orienterade på ett sådant sätt att den polära delen av molekylen är vänd mot lösningsmedlet, medan den opolära delen trycks in i gasfasen.
I Shishkovskys ekvation  , är den konstant för den homologa serien.
, är den konstant för den homologa serien.
Ytaktiv handling börjar dyka upp med n>5. Vid koncentrationer högre än koncentrationen av det monomolekylära skiktet sker micellisering i ytaktiva lösningar.
Micelle- aggregatet av amfifila ytaktiva molekyler kallas, vars kolväteradikaler bildar kärnan, och de polära grupperna omvandlas till vattenfasen.
Micellmassa - micellmassa.
H  antalet molekyler är antalet aggregation.
antalet molekyler är antalet aggregation.
Sfäriska miceller
Vid micellisering upprättas en jämvikt i lösningen
CMC är den kritiska micellkoncentrationen.
Eftersom vi anser att micellen är en separat fas:
För den homologiska serien finns det en empirisk ekvation:
aär den funktionella gruppens upplösningsenergi.
b är adsorptionspotentialökningen, arbetet med adsorption per metylenenhet.
är adsorptionspotentialökningen, arbetet med adsorption per metylenenhet.
Närvaron av en kolvätekärna i miceller gör det möjligt för föreningar som är olösliga i vatten att lösa sig i vattenlösningar av ytaktiva ämnen, detta fenomen kallas solubilisering (det som löser sig är ett solubilisat, ytaktivt ämne är ett solubiliseringsmedel).
Slammet kan vara helt opolärt, kan innehålla både polära och opolära delar och kommer att vara orienterat som en ytaktiv molekyl.
I vilket fall som helst, under solubilisering, sker en ökning av micellmassan och aggregationstalet inte bara på grund av inkluderingen av solubilisatet, utan också på grund av en ökning av antalet ytaktiva molekyler som är nödvändiga för att upprätthålla jämviktstillståndet.
Solubilisering är desto effektivare desto lägre molekylvikt har solubilisatet.

 ~ 72 mN/m.
~ 72 mN/m.
 ~ 33 mN/m.
~ 33 mN/m.
Effektiviteten av ytaktiva ämnen beror på storleken på CMC.
2D Ytskiktstryck

→ -krafter av ytspänning.
- tvådimensionellt tryck.
Ytskiktet är en kraft lika med skillnaden mellan ytspänningarna för en lösning av ytaktivt medel och ett rent lösningsmedel, riktad mot en ren yta.
En jämvikt upprättas mellan lösningen och ytskiktet



På  det finns ett område där
det finns ett område där  linjärt beroende av koncentration.
linjärt beroende av koncentration.
G [mol/m2].
 område som upptas av en mol av ett ämne
område som upptas av en mol av ett ämne
Då kommer den tvådimensionella tryckisotermen att ha formen 
 är den tvådimensionella tryckisotermen.
är den tvådimensionella tryckisotermen.
Missbruk  från S M:
från S M:

På  - det tvådimensionella trycket ökar kraftigt. På
- det tvådimensionella trycket ökar kraftigt. På  tvådimensionell är deformerad, vilket orsakar en kraftig tillväxt
tvådimensionell är deformerad, vilket orsakar en kraftig tillväxt  .
.
Filmen på båda sidor som begränsas av samma faser kallas dubbelsidig. I sådana filmer observeras en konstant rörelse av moderluten.
Filmer som är mindre än 5 nm tjocka kallas svarta filmer.

Adsorptionsskikt bör ha två egenskaper: viskositet och lätt rörlighet, flytbarhet och elasticitet.
Marangoni-effekten är självläkande.
Gibbs triangel,  - övertryck.
- övertryck.
Filmen sträcks och på grund av att en del av vätskan är borta rusar de ytaktiva medlen in i det fria utrymmet. Gibbs triangel.
Effekt av kroppars adsorptionsstyrka.
Det finns alltid ett adsorptionsskikt på filmytan, för vilket då
Langmuirs ekvation:


 till tvådimensionellt tryck
till tvådimensionellt tryck
 - analog till Shishkovsky-ekvationen
- analog till Shishkovsky-ekvationen
elektrokinetiska fenomen. Dubbelt elektriskt lager (DES).
Helemholtz modell. Gouy-Chapman teori.
1808 flygning

U – formade rör, nedsänkt i det 2 elektroder. Lagen om kommunicerande kärl bryts och det finns en förändring i vätskenivån i röret - elektrokinetiska fenomen.
Kinetiska fenomen:
elektrofores
Elektroosmos
Flöde (flöde) potential
Sedimentationspotential
1 och 2 uppstår när en potentialskillnad appliceras, 3 och 4 orsakar stansningen och sedimenteringen av kolloidala partiklar uppkomsten av en potentialskillnad.
Elektroosmos är rörelsen av ett dispersionsmedium i förhållande till en stationär dispergerad fas under inverkan av en elektrisk ström.
elektrofores är rörelsen av partiklar i den dispergerade fasen i förhållande till ett stationärt dispersionsmedium under inverkan av en elektrisk ström.
P  Orsaken till förekomsten av elektrokinetiska fenomen är den rumsliga separationen av laddningar och uppkomsten av ett dubbelt elektriskt lager.
Orsaken till förekomsten av elektrokinetiska fenomen är den rumsliga separationen av laddningar och uppkomsten av ett dubbelt elektriskt lager.
Det elektriska dubbelskiktet är en platt kondensator, en platta är bildad av potentialbestämmande joner, den andra av motinoer. Jonerna är också förorenade när potentialbestämmande samjoner trycks in i huvuddelen av lösningen. Avstånd mellan plattorna  . Potentialen faller linjärt, potentialskillnaden
. Potentialen faller linjärt, potentialskillnaden  .
.
En extern potentialskillnad orsakar uppkomsten av en skjuvmodul  är ett par krafter per ytenhet som verkar längs ytan av en fast kropp.
är ett par krafter per ytenhet som verkar längs ytan av en fast kropp.

Vid jämvikt är skjuvmodulen lika med den viskösa friktionsmodulen (  ).
).

I våra förhållanden  ,
,

 - Helemholtz-Smalukovsky ekvation
- Helemholtz-Smalukovsky ekvation
 - linjär hastighetsförskjutning i faser.
- linjär hastighetsförskjutning i faser.
Eär det elektriska fältets styrka.
 - potentialskillnad mellan plattorna
- potentialskillnad mellan plattorna
 - elektroforetisk rörlighet [m2/(V*s)].
- elektroforetisk rörlighet [m2/(V*s)].
Helemholtz-modellen tar inte hänsyn till molekylers termiska rörelse. I verkligheten är fördelningen av joner i dubbelskiktet mer komplex.
Gouy och Chapman identifierade följande orsaker till DES:
Övergången av en jon från en fas till en annan när jämvikt är etablerad.
Jonisering av fast fasmaterial.
Komplettering av ytan av joner som finns i dispersionsmediet.
Polarisering från en extern strömkälla.
Det elektriska dubbelskiktet har en suddig eller diffus struktur. Joner tenderar att vara jämnt fördelade i det diffusa skiktet.
Det diffusa skiktet består av motjoner, längden på skiktet bestäms av deras kinetiska energi. Vid en temperatur som tenderar till absolut noll är motinoer så nära potentialbestämmande joner som möjligt.
Denna teori bygger på två ekvationer:
Boltzmanns ekvation

 - arbeta mot krafterna från elektrostatisk interaktion.
- arbeta mot krafterna från elektrostatisk interaktion.
 är bulkladdningstätheten.
är bulkladdningstätheten.

Poissons ekvation 
Eftersom DEL-tjockleken är mycket mindre än partikelstorleken, och för en platt DEL, är derivatan med avseende på koordinater  och
och  är avskaffad.
är avskaffad. 
För e y med y<<1 функцию можно разложить в ряд Маклорена:

Vi begränsar oss till två medlemmar i serien, då:


 - DEL-tjocklek är det avstånd från vilket DEL-potentialen minskar e en gång.
- DEL-tjocklek är det avstånd från vilket DEL-potentialen minskar e en gång.
Ju lägre temperatur, desto mindre  . Vid Т→0 – platt DES. Ju högre koncentration, desto mer jag, desto mindre
. Vid Т→0 – platt DES. Ju högre koncentration, desto mer jag, desto mindre  .
.





 "–" betyder att potentialen minskar med avståndet. =>
"–" betyder att potentialen minskar med avståndet. =>
=>

 ,
,
 - potentialen minskar exponentiellt.
- potentialen minskar exponentiellt.
Potential för ytladdningstäthet: 
Ytladdning är en rymdladdning med motsatt tecken, integrerad över avstånd.




=>


Där potentialen minskar med 2,7 gånger - 
Dubbla lagerkapacitet 
Nackdelen med teorin är att närvaron av Helemholtz-skiktet inte beaktas, d.v.s. tar inte hänsyn till  , därav felen vid bestämning av huvudparametrarna. Det förklarar inte heller effekten av joner av olika natur på tjockleken av det elektriska dubbelskiktet.
, därav felen vid bestämning av huvudparametrarna. Det förklarar inte heller effekten av joner av olika natur på tjockleken av det elektriska dubbelskiktet.
Sterns teori. Strukturen av en kolloidal micell.
Det elektriska dubbelskiktet består av två delar: tätt och diffust. Ett tätt skikt bildas som ett resultat av interaktionen av potentialbildande joner med specifikt adsorberade. Dessa joner är som regel delvis eller helt uttorkade och kan ha antingen samma eller motsatt laddning till de potentialbestämmande jonerna. Det beror på förhållandet mellan energin för elektrostatisk interaktion  och specifik adsorptionspotential
och specifik adsorptionspotential  . Jonerna i det täta lagret är fixerade. Den andra delen av jonerna ligger i det diffusa lagret, dessa joner är fria och kan röra sig djupt in i lösningen, d.v.s. från ett område med högre koncentration till ett område med lägre koncentration. Den totala laddningstätheten består av två delar.
. Jonerna i det täta lagret är fixerade. Den andra delen av jonerna ligger i det diffusa lagret, dessa joner är fria och kan röra sig djupt in i lösningen, d.v.s. från ett område med högre koncentration till ett område med lägre koncentration. Den totala laddningstätheten består av två delar.

 - Helmholtz lagerladdning
- Helmholtz lagerladdning
 -Diffus lagerladdning
-Diffus lagerladdning
Ytan har ett visst antal adsorptionscentra, som var och en interagerar med en motjon. Konstanten för en sådan kvasikemisk reaktion är:

 , var
, var  - molfraktion av motjoner i lösning
- molfraktion av motjoner i lösning
Helmholtz distribution
Potentialen minskar linjärt
Gouy potentialdistribution. Det finns inget tätt lager, potentialen minskar exponentiellt från värdet 
Sternfördelning.
Till en början är den potentiella minskningen linjär och sedan exponentiellt.
När ett elektriskt fält appliceras vid elektrofores är det inte partikeln i den fasta fasen som rör sig direkt, utan partikeln i den fasta fasen med ett lager av omgivande joner. DES upprepar formen av partikeln i den dispergerade fasen. När en potential appliceras rivs en del av det diffusa lagret av. Brytlinjen kallas glidande gräns.
Potentialen som uppstår vid glidgränsen till följd av att en del av det diffusa skiktet lossnar kallas elektrokinetisk potential(Zeta potential  ).
).
En partikel av en dispergerad fas, med ett lager av motjoner som omger den och ett dubbelt elektriskt lager, kallas micell.
Regler för att skriva kolloidala miceller:


1-1 laddningselektrolyt
T är en partikel av den dispergerade fasen.
AA är gränsen mellan de täta och diffusa delarna.
BB är glidgränsen.
Slirgränsen kan eller kanske inte sammanfaller med linje AA.
pH-värdet vid vilket zetapotentialen är noll kallas isoelektrisk punkt.
CaCl2 + Na2SO4 → CaSO4 ↓ + 2NaCl
1. I överskott av CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n-x)Cl-) 2 x + x Cl - - rekordmiceller.
CaSO 4 m - aggregat.
CaSO 4 m∙nCa 2+ är kärnan.
CaSO 4 m∙nCa 2+ 2( n-x)Cl - partikel.
2. I överskott av Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO4 m∙nSO4 2- 2(n-x)Na+) 2x- 2xNa+ - micell
CaSO 4 m - aggregat.
CaSO 4 m∙nSO 4 2 + är kärnan.
CaS04 m∙nSO4 2-2(n-x)Na+-partikel
Helemholtz-Smoluchowskis ekvation

 - linjär hastighet för förskjutning av gränser (vid elektroosmos).
- linjär hastighet för förskjutning av gränser (vid elektroosmos).
 - potentialskillnad på kondensatorplattorna (vid elektroosmos).
- potentialskillnad på kondensatorplattorna (vid elektroosmos).


 - lösningens volymetriska flödeshastighet, Sär cellens tvärsnittsarea.
- lösningens volymetriska flödeshastighet, Sär cellens tvärsnittsarea.

Eär det elektriska fältets styrka.


 (för elektroosmos).
(för elektroosmos).
För flödespotentialen:

 - potential
- potential
 - membrantryck
- membrantryck
Som regel är värdet av elektroforetiska rörligheter och elektroosmotiska rörligheter mindre än de beräknade. Detta beror på:
Avslappningseffekt (under rörelsen av en partikel av den dispergerade fasen, kränks symmetrin i den joniska atmosfären).
Elektroforetisk bromsning (förekomsten av ytterligare friktion som ett resultat av rörelsen av motjoner).
Förvrängning av strömlinjer vid elektriskt ledande partiklar.
Samband mellan ytspänning och potential. Lippmanns ekvation.
Bildningen av DEL sker spontant på grund av systemets önskan att minska sin ytenergi. I konstanssammanhang T och sid den generaliserade ekvationen för termodynamikens första och andra lag ser ut så här:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1:a Lippmann-ekvationen.
- 1:a Lippmann-ekvationen.
 är ytladdningstätheten.
är ytladdningstätheten.
 - differentiell kapacitans.
- differentiell kapacitans.
 - 2:a Lippmann-ekvationen.
- 2:a Lippmann-ekvationen.
Med- kapacitet.
Vi löser den första Lippmann-ekvationen och den fundamentala adsorptionsekvationen:
 ,
,


 , då
, då
 - Nernsts ekvation
- Nernsts ekvation
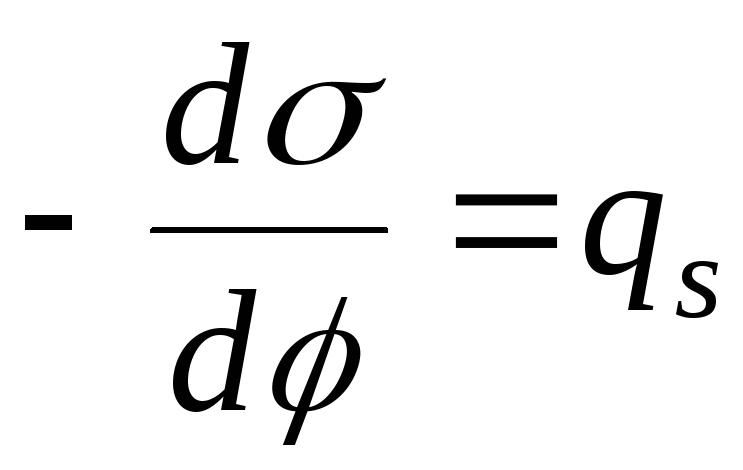 ,
,
 ,
,




 - ekvationen för den elektrokapillära kurvan (ECC).
- ekvationen för den elektrokapillära kurvan (ECC).
PÅ  :
: , men
, men 
Katjoniska ytaktiva ämnen (CSAS) reducerar den katodiska grenen av ECC.
Anjoniska ytaktiva ämnen (ASS) reducerar den anodiska grenen av ECC.
Nonjoniska ytaktiva ämnen (NSA) reducerar mittdelen av ECC.
Stabilitet av spridda system. Kiltryck.
Dispergerade system kan delas in:
Termodynamiskt instabila system kan vara kinetiskt stabila på grund av övergången till ett metastabilt tillstånd.
Det finns två typer av stabilitet:
Sedimentationsstabilitet (med hänsyn till gravitation).
Aggregativ stabilitet. (i förhållande till att sticka)
Koaguleringär processen där partiklar klibbar ihop, vilket leder till förlust av aggregativ stabilitet. Koagulering kan orsakas av förändringar i temperatur, pH, omrörning, ultraljud.
Särskilj koagulation:
Reversibel.
Irreversibel.
Koaguleringen fortsätter med införandet av elektrolyter.
Koaguleringsregler:

Filma– Det här är den del av systemet som ligger mellan två gränssnitt.
upplösande tryck uppstår med en kraftig minskning av filmtjockleken som ett resultat av interaktionen av närmande ytskikt.

«-» - när filmtjockleken minskar, ökar det lösgörande trycket.
P 0 är trycket i bulkfasen, som är en fortsättning på mellanskiktet.
P 1 är trycket i filmen.
Teori om stabilitet. DLFO (Deryagin, Landau, Fairway, Overbeck).
Enligt DLVO-teorin särskiljs två komponenter i det sönderfallande trycket:
elektrostatisk PE (positiv, det beror på krafterna från elektrostatisk repulsion). Motsvarar en minskning av Gibbs energi med ökande filmtjocklek.
Molekyl PM (negativ, på grund av attraherande krafters verkan). Det orsakas av komprimeringen av filmen på grund av kemiska ytkrafter, krafternas verkningsradie är tiondels nm med en energi av storleksordningen 400 kJ/mol.
Total interaktionsenergi:

 - systemet är aggregerat stabilt
- systemet är aggregerat stabilt
 - instabilt system
- instabilt system
P  positiv komponent.
positiv komponent.
Ökningen beror på ökningen av potentiell energi under komprimering av tunna filmer. För tjocka filmer kompenseras överskottet av jonenergi och är lika med energiinteraktionen i huvuddelen av dispersionsmediet.
Om en  (
( - film tjocklek,
- film tjocklek,  - jonens radie) förtunning av filmen leder till att molekyler och joner i den försvinner och reduceras med minimal ytenergi. Antalet intilliggande partiklar minskar, vilket gör att den potentiella energin hos de partiklar som finns kvar i filmen ökar.
- jonens radie) förtunning av filmen leder till att molekyler och joner i den försvinner och reduceras med minimal ytenergi. Antalet intilliggande partiklar minskar, vilket gör att den potentiella energin hos de partiklar som finns kvar i filmen ökar.


DLVO-teorin betraktar interaktionen mellan partiklar som interaktionen mellan plattor.

Partiklar interagerar inte



 - Laplace ekvation,
- Laplace ekvation,  ,
,


För svagt laddade ytor
För mycket laddade ytor:

Den molekylära komponenten är interaktionen mellan två atomer:
 ~
~
Interaktion mellan en atom och en yta:

Låt oss ta två rekord:
D  För att erhålla den molekylära komponenten är det nödvändigt att summera alla interaktionsenergier för atomerna på den högra och vänstra plattan.
För att erhålla den molekylära komponenten är det nödvändigt att summera alla interaktionsenergier för atomerna på den högra och vänstra plattan.
var  - Hamakers konstant (tar hänsyn till naturen hos interagerande kroppar).
- Hamakers konstant (tar hänsyn till naturen hos interagerande kroppar).
Det där. interaktionsenergin för partiklar i ett system kan uttryckas med hjälp av potentialkurvor.
I är det primära potentiella minimumet. Detta är en zon av irreversibel koagulering, attraktionskrafterna råder.
II - zon av aggregativ stabilitet, frånstötande krafter råder.
III - sekundär potentialminimum (eller flockningszon). Mellan partiklarna i den dispergerade fasen finns ett elektrolytskikt, och partiklarna kan separeras och överföras till zonen med aggregativ stabilitet.

Kurva 1 – systemet är aggregerat stabilt.
Kurva 2 är stabil i zon I, inte stabil i zon II.
Kurva 3 - koagulering inträffade i systemet.
Kurva 4 - vid punkt 4, den totala energin för interaktion U=0,  , motsvarar denna extrema punkt början av snabb koagulering.
, motsvarar denna extrema punkt början av snabb koagulering.
Det finns två fall:
1. Ytor är svagt laddade:
U \u003d U E + U M \u003d 0
 (1)
(1)
2)

 (2)
(2)



 - detta är tjockleken på lagret som motsvarar början av koaguleringsprocessen.
- detta är tjockleken på lagret som motsvarar början av koaguleringsprocessen.






 - för svagt laddade ytor
- för svagt laddade ytor
 sedan
sedan 
2. För mycket laddade ytor:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Låt oss kvadrera (3)

Koagulering:
Vid specifik adsorption kan joner adsorberas i en superekvivalent mängd på ett sådant sätt att ytan kan ändra sin laddning. Ytan laddas om.
I fallet med specifik adsorption kan inte bara joner med motsatta tecken, utan även av en jon adsorberas.
Om joner med samma tecken som ytan adsorberas, kommer det inte att finnas en droppe i potentialen i ytskiktet, utan dess tillväxt.

Neutraliseringskoagulering (förekommer med deltagande av svagt laddade partiklar och beror inte bara på laddningen av den koagulerande elektrolyten, utan också på potentialen vid gränsen för de täta och diffusa skikten).
Smoluchowskis teori om snabb koagulering.

Beroende av koagulationshastighet på elektrolytkoncentration.
I – koagulationshastigheten är låg,
II - koagulationshastigheten är praktiskt taget proportionell mot elektrolytkoncentrationen.
III - området för snabb koagulering, hastigheten är praktiskt taget oberoende av koncentrationen.
Nyckelord:
Den initiala solen är monodispers, liknande partiklar har en sfärisk form.
Alla partikelkollisioner är effektiva.
När två primära partiklar kolliderar bildas en sekundär partikel. Sekundär + primär = tertiär. Primär, sekundär, tertiär - mångfald.
När det gäller kemisk kinetik kan koaguleringsprocessen beskrivas med ekvationen:

Lösningen blir ekvationen: 
 - tid för halvkoagulering. Detta är den tid under vilken antalet solpartiklar minskar med 2 gånger.
- tid för halvkoagulering. Detta är den tid under vilken antalet solpartiklar minskar med 2 gånger.

 ,
,
 ,
,
 ,
,


När multipliciteten ökar skiftar koagulationskurvornas maximum mot större värden  .
.
Nackdelar:
Antagande om monodispersitet.
Antagandet om effektiviteten av alla kollisioner.
Vi rekommenderar också
 Produktivt och reproduktivt tänkande
Produktivt och reproduktivt tänkande
 Rimlig egoism - vad är teorin om rimlig egoism?
Rimlig egoism - vad är teorin om rimlig egoism?
 Boris Nikolajevitj Jeltsin, Rysslands första president
Boris Nikolajevitj Jeltsin, Rysslands första president
 Underjordiska slagsmål. Underjordiska kungar. Vad är "att slåss inte för massorna"? Var kan man slåss om pengar?
Underjordiska slagsmål. Underjordiska kungar. Vad är "att slåss inte för massorna"? Var kan man slåss om pengar?
 Yakov Pavlov och andra hjältar från Stalingrad du behöver veta
Yakov Pavlov och andra hjältar från Stalingrad du behöver veta
 Överlev en olycka till sjöss i en dröm - upplev i verkligheten en ny kärlek
Överlev en olycka till sjöss i en dröm - upplev i verkligheten en ny kärlek