Thermodynamique du processus d'adsorption. Thermodynamique des processus d'adsorption Modifications des fonctions thermodynamiques au cours de l'adsorption
L'adsorption sous forme de concentration spontanée de molécules à la surface s'accompagne d'une diminution de l'entropie du système. Comme le critère de spontanéité du processus est
∆N - T · ∆S = ∆G< 0,
alors l'adsorption n'est possible qu'à ∆Н< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. Avec une augmentation de la température, l'équilibre se déplace vers le processus endothermique, c'est-à-dire la désorption.
Adsorption sur une surface solide
1. adsorption monomoléculaire.
Selon la théorie de Langmuir, les molécules d'adsorbat interagissent avec la surface de l'adsorbant, formant finalement une couche monomoléculaire. Dans ce cas, le degré de remplissage () de la surface avec la substance adsorbée lors de l'adsorption à partir de la phase gazeuse
du liquide
où K est la constante d'équilibre (constante d'adsorption);
p est la pression partielle du gaz adsorbé ;
c est la concentration de la substance adsorbée.
La dépendance de β sur p (ou c) est représentée par un graphique (isotherme d'adsorption, Т = const) sur la Fig. 1. 1.3.

Riz. 1.3. Degré de remplissage de la surface avec la substance adsorbée
Aux faibles concentrations et pressions partielles, l'adsorption est proportionnelle à la concentration ou à la pression partielle :
R<< 1, β ≈ К· r oris<< 1, β ≈ К· s, c'est-à-dire la section initiale de l'isotherme est approximativement linéaire et tan α \u003d K (tg α est déterminé par la pente de la courbe à p (ou c) → 0: ou ).
Si - le nombre de moles de substance adsorbée pour 1 g d'adsorbant; - le nombre maximal possible de moles de substance adsorbée pour 1 g d'adsorbant ("capacité monocouche"), puis
En remplaçant β dans l'équation (1.3) (pour le cas de l'adsorption à partir de la phase gazeuse, la concentration Avec dans les équations doit être remplacé par la pression R), on a:
 (1.6)
(1.6)
Puisque et K dans ce couple adsorbant-adsorbant sont des constantes (à J= const), alors la dépendance peut être trouvée et À(Fig. 1.4).

Riz. 1.4. Solution graphique de l'équation d'adsorption
obtenu en extrapolant la dépendance linéaire expérimentale à () = 0 ; et, puisque , alors , .
La valeur peut être utilisée pour déterminer la surface spécifique de l'adsorbant UD (en m 2 pour 1 g d'adsorbant), si l'aire ω occupée en surface par une molécule d'adsorbant est connue (déterminée à partir de la taille de la molécule) :
UD = · ω · Na, (1.7)
où Na est le nombre d'Avogadro (Na = 6,02 10 23).
À son tour, la valeur connue de SD peut être utilisée pour calculer ou ω de n'importe quelle substance par son adsorption sur un adsorbant donné.
2. Adsorption polymoléculaire.
L'équation (1.5) décrit une courbe avec saturation, c'est-à-dire à
p (ou c) → ∞ tend vers la valeur limite égale à (Fig. 1.5, a).

Fig.1.5. Isothermes d'adsorption :
a – adsorption à saturation ; b – adsorption polymoléculaire
Cependant, dans certains cas, les isothermes d'adsorption ressemblent à celles illustrées à la Fig. 1.5b, c'est-à-dire n'atteint pas la limite même à haut p (ou c).
Les dépendances du type illustré à la fig. 1.5b correspondent à une adsorption polymoléculaire. En règle générale, ces isothermes sont caractéristiques de substances à fortes interactions intermoléculaires (par exemple, l'eau). Lorsque les centres d'adsorption à la surface de l'adsorbant sont occupés (la couche monomoléculaire est saturée), "l'atterrissage" des molécules d'adsorbat suivantes se produit en raison d'interactions intermoléculaires avec des molécules déjà adsorbées (Fig. 1.6). La chaleur d'une telle adsorption est proche en valeur absolue, mais de signe opposé, de la chaleur d'évaporation du liquide correspondant (pensez pourquoi).

Fig.1.6. Schéma d'adsorption :
a - adsorption monomoléculaire ; b - adsorption polymoléculaire
Alors que tu te rapproches Rà la pression de vapeur saturante de la substance adsorbée, elle commence à se condenser à la surface de l'adsorbant ; en conséquence, elle augmente rapidement avec l'augmentation R.
Thermodynamique des procédés d'adsorption.
| Le nom du paramètre | Sens |
| Sujet de l'article : | Thermodynamique des procédés d'adsorption. |
| Rubrique (catégorie thématique) | Éducation |
Définitions de base et méthodes de classification des processus d'adsorption.
L'adsorption fait référence à des phénomènes se produisant à la suite d'une diminution spontanée de l'énergie de surface.
Adsorption- le processus de redistribution spontanée réversible ou irréversible des composants d'un système hétérogène entre la couche superficielle et le volume d'une phase homogène.
Dans les systèmes multicomposants, le composant qui abaisse la tension interfaciale est préférable à la couche de surface. Dans les systèmes monocomposants, lors de la formation de la couche superficielle, sa structure change (une certaine orientation des atomes et des molécules, polarisation), appelée autoadsorption.
La phase la plus dense sur laquelle se localisent les interactions d'adsorption est appelée adsorbant. La substance redistribuée entre le volume de la phase homogène et la couche superficielle est désignée par le terme ʼʼ adsorberʼʼ.
Dans certains cas, le processus d'adsorption est réversible. Dans ce cas, sous certaines conditions, une partie des molécules adsorbées, du fait de phénomènes de cinétique moléculaire, peut passer de la couche superficielle dans le volume de la phase. Le processus inverse d'adsorption est appelé désorption.
Méthodes de classification des procédés d'adsorption.
Classification des processus d'adsorption selon l'état d'agrégation des phases en interaction. Compte tenu de la dépendance à l'état agrégé des phases adjacentes, on distingue les types de processus d'adsorption suivants:
Adsorption de gaz sur adsorbants solides ;
Adsorption des substances dissoutes aux interfaces ʼʼsolide-liquideʼʼ et ʼʼliquide-liquideʼʼ ;
Adsorption de tensioactifs à l'interface ʼʼliquide-gazʼʼ.
Classification des processus d'adsorption selon le mécanisme d'interaction de l'adsorbant et de l'adsorbat. L'adsorption peut être considérée comme l'interaction des molécules d'adsorbat avec les centres actifs de l'adsorbant. Selon le mécanisme de leur interaction, les types d'adsorption suivants sont subdivisés :
1) adsorption physique (moléculaire)- l'interaction entre les molécules de l'adsorbat et l'adsorbant s'effectue grâce aux forces de van der Waals, liaisons hydrogène (sans apparition de réactions chimiques) ;
2) adsorption chimique (chimisorption)- la fixation des molécules d'adsorbat sur les centres actifs de l'adsorbant se produit à la suite de réactions chimiques de divers types (à l'exception des réactions d'échange d'ions) ;
3) adsorption échangeuse d'ions (échange d'ions) - la redistribution de la substance adsorbée entre la solution et la phase solide (échangeur d'ions) selon le mécanisme des réactions d'échange d'ions.
Pour une description quantitative des processus d'adsorption, deux grandeurs sont utilisées.
1) Adsorption absolue est la quantité (mole) ou la masse (kg) de l'adsorbat par unité de surface ou de masse de l'adsorbant. Désignation - A ; unité : mol/m 2 , mol/kg, kg/m 2 , kg/kᴦ.
2) Adsorption de Gibbs (excès)- un excès de substance adsorbée dans une couche superficielle d'une certaine épaisseur par rapport à sa quantité dans le volume d'une phase homogène, rapportée à une unité de surface ou masse d'adsorbant. Désignation - G ; unité : mol/m 2 , mol/kᴦ.
La relation entre l'adsorption absolue et l'excès d'adsorption peut être illustrée à l'aide de l'équation :
G \u003d A - c * h (3.1)
où c est la concentration à l'équilibre de la substance dans le volume de la phase, mol/m3 ;
h est l'épaisseur de la couche superficielle, prise conditionnellement égale à 10 -9 m.
Dans les systèmes hétérogènes multicomposants, lorsque l'un ou l'autre composant est redistribué entre le volume d'une phase homogène et la couche superficielle, l'équation de l'excès d'énergie interne de la surface est valable :
U = T * S + s * s + Sm je * n je (3.2)
En ramenant tous les termes de l'équation à l'aire unitaire de la surface interfaciale, on obtient :
U s = T * S s + s + Sm je * Ã je (3.3)
où Г i = n i / s est l'excès du i-ème composant dans la couche de surface, c'est-à-dire l'adsorption de Gibbs.
Pour un système à un composant, l'équation (3.3) prend la forme :
G s = s + m * Ã (3.4)
où G s = U s - T * S s est l'énergie de Gibbs de la surface ou le travail de création d'une unité de surface de la surface;
m * Г - compactage de la substance de la substance adsorbée dans la couche de surface.
Sur la base de l'équation (3.4), nous pouvons conclure que lors de l'adsorption, le travail de création d'une surface interfaciale consiste en un travail de formation d'une surface (rupture des liaisons cohésives dans la masse de la phase d'adsorbat) et de compactage de la substance dans la couche de surface.
Dans l'état d'équilibre dynamique entre l'adsorbant et l'adsorbat, la variation de l'énergie de Gibbs d'un système hétérogène ΔG = 0, la thermodynamique du processus d'adsorption est décrite par une équation appelée Équation fondamentale d'adsorption de Gibbs:
Ds = SГ i * dm i (3.5)
Cette équation est universelle, puisqu'elle est valable pour tous les types de procédés d'adsorption
Cas particuliers de l'équation d'adsorption de Gibbs.
1) Adsorption à partir de solutions.
Pour le potentiel chimique du i-ème composant du système lors de l'adsorption aux interfaces "liquide - adsorbant solide" et "liquide - gaz", les équations sont valables :
m je = m je 0 + R*T*ln une je (3.6)
dm je = R*T* d ln une je (3.7)
où m i 0 est le potentiel chimique du i-ème composant du système dans des conditions standard ;
a i - activité du i-ème composant du système dans des conditions standard.
Sur cette base, l'équation d'adsorption de Gibbs prendra la forme :
à je = - une je / R*T * (ds / da je) (3.8)
Pour les solutions non électrolytiques, on prend a i \u003d c i, alors :
Г je \u003d - s / R * T * (ds / ds) (3.9)
Pour les solutions électrolytiques :
à i = - ñ ± n / R*T * (ds / dñ ± n) (3.10)
où c ± est la concentration ionique moyenne de la solution ;
n est le coefficient stoechiométrique.
2) Adsorption de substances de la phase gazeuse.
Conformément à l'équation de Mendel-eev-Claiperon :
P \u003d c * R * T (3.11)
A cet égard, l'équation de Gibbs pour l'adsorption des gaz sur les adsorbants solides s'écrit sous la forme suivante :
à i = - Р / R*T * (ds / dР) (3.12)
En pratique, l'équation d'adsorption de Gibbs permet, en fonction de la mesure de la tension superficielle à différentes valeurs de concentration de liquide ou de pression de gaz d'équilibre, de calculer la quantité d'adsorption de substances dans la couche interfaciale, pour laquelle la tension superficielle est déterminée.
Thermodynamique des procédés d'adsorption. - concepts et types. Classification et caractéristiques de la catégorie "Thermodynamique des processus d'adsorption". 2017, 2018.
Page courante : 6 (le livre compte 19 pages au total) [extrait de lecture accessible : 13 pages]
Police de caractère:
100% +
34. Nature des forces d'adsorption
L'interaction entre les molécules de l'adsorbant avec la surface de l'adsorbant à la soi-disant. l'adsorption physique peut être due à diverses raisons. Ensuite, le potentiel qui détermine l'interaction d'une molécule d'adsorbant avec un atome d'un adsorbant non polaire peut être exprimé comme suit :
θ = −Cr 6 +BR 12 ,
où r est la distance entre les centres des particules ; C est la constante d'attraction de la dispersion ; B est une constante qui caractérise l'énergie des forces répulsives.
Il est bien évident qu'à des distances relativement éloignées, les forces attractives doivent prévaloir, et à des distances proches, les forces répulsives. Aussi, à certaines distances, ces forces doivent être égales, ce qui correspondra à l'énergie libre minimale. Mais il est important de noter que lors de l'adsorption, les forces de dispersion agissent simultanément entre chaque particule non polaire.
Comme l'énergie d'interaction des particules peut rapidement décroître avec la distance, il suffit d'effectuer une sommation sur les atomes adsorbants les plus proches pour déterminer le potentiel des forces d'adsorption. Il est important que, dans le cas de l'adsorption de molécules non polaires complexes, l'énergie potentielle puisse être approximativement calculée comme la somme de toutes les énergies potentielles d'adsorption des unités de la molécule.
Si l'adsorbant est constitué d'ions, l'action des forces de dispersion déjà connues peut être complétée par l'action des forces d'induction d'attraction des dipôles, qui sont induites dans les molécules de l'adsorbant par un champ électrique, qui, à son tour, est créé par les ions du réseau adsorbant.
Avec une telle interaction, la part des forces inductives dans l'interaction d'adsorption peut être proportionnelle à la polarisabilité de la molécule adsorbante et au carré de l'intensité du champ sur cette surface adsorbante.
Si, au contraire, des molécules d'adsorbant polaire sont adsorbées sur un adsorbant polaire, alors les dipôles dans ce cas polarisent les atomes de l'adsorbant, c'est-à-dire comme s'ils induisaient des moments électriques en eux. En raison de cette influence, l'interaction inductive s'ajoute à celle de dispersion.
L'interaction inductive elle-même est généralement faible et, en fonction du dipôle de la molécule adsorbante et de la polarisabilité de l'adsorbant, peut atteindre de grandes valeurs. Dans le cas où des molécules sont adsorbées sur un adsorbant qui a des ions ou des dipôles à la surface, un soi-disant. interaction des ions ou dipôles de l'adsorbant avec le champ électrostatique de l'adsorbant lui-même.
Dans ce cas, les molécules adsorbantes peuvent même s'orienter dans le champ de l'adsorbant, et il se produit une interaction coulombienne orientationnelle. Il arrive généralement que les énergies des interactions inductives et orientationnelles soient inférieures à l'énergie de l'interaction de dispersion, et on suppose donc que l'énergie d'attraction intermoléculaire est déterminée par l'énergie d'attraction de dispersion.
De plus, la formation d'une liaison hydrogène peut servir de cause d'adsorption. Une liaison de ce type peut apparaître lors de l'adsorption sur des adsorbants contenant des groupes hydroxyle de molécules telles que l'eau, les alcools, l'ammoniac et les amines à la surface. Lorsqu'une liaison hydrogène est formée, l'énergie d'interaction de l'adsorbant avec l'adsorbant peut être assez grande et la chaleur dégagée lors d'une telle adsorption est bien supérieure à la chaleur d'adsorption de substances de forme et de taille de molécules similaires, mais ne forment pas de liaison hydrogène.
Il est important de noter que, connaissant la description thermodynamique de la couche superficielle à la frontière "adsorbant - adsorbant", sa structure, la nature des différents types de forces, la dynamique du processus, on peut procéder à l'étude de processus plus complexes processus d'adsorption.
35. Adsorption sous forme de concentration spontanée sur l'interface de phase de substances qui réduisent la tension interfaciale
Les tensioactifs sont divisés en deux grands groupes : actif et inactif substances.
Les tensioactifs peuvent s'accumuler dans la couche superficielle et, dans ce cas, une adsorption positive se produit. g > 0.
Ces types de substances doivent avoir une tension superficielle qui, à son tour, doit être inférieure à la tension superficielle du solvant, sinon l'accumulation de la substance dans la couche superficielle serait défavorable, et doit avoir une solubilité relativement faible. Avec une solubilité suffisamment bonne, les molécules de tensioactif ont tendance à quitter la surface profondément dans la solution. Par conséquent, les tensioactifs seront préférentiellement poussés hors de la masse du liquide vers la surface.
Mais avec l'accumulation de substances à la limite de la solution dans les molécules de ces substances, qui interagissent faiblement les unes avec les autres, l'interaction intermoléculaire dans la couche superficielle diminuera et la tension superficielle diminuera.
Tensioactifs par rapport à la couche d'eau, il existe de nombreux types de composés organiques, des acides gras avec un radical hydrocarboné suffisamment grand, des sels de ces acides (savons), des acides sulfoniques et leurs sels, ainsi que divers types d'alcools et d'amines. Un trait caractéristique de la plupart des molécules est leur amphiphilie : la molécule est constituée de deux parties d'un groupe polaire et d'un radical hydrocarboné non polaire. Avoir un moment dipolaire significatif et un groupe polaire bien hydratant peut déterminer l'affinité du tensioactif pour le milieu aqueux. Mais le radical hydrocarbure est la cause qui abaisse la solubilité de ces composés.
Agents de surface inactifs- ces types de substances, ayant tendance à quitter la surface du liquide dans son volume, en conséquence, le soi-disant. adsorption négative g < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Substances inactives en surface par rapport à l'eau, il existe de nombreux électrolytes inorganiques: acides, alcalis, sels. Les molécules de substances inactives en surface n'ont pas de partie hydrophobe et peuvent se décomposer dans l'eau en ions hautement hydratants.
Exemples les substances inactives en surface sont également des composés organiques dans lesquels la partie non polaire de la molécule est absente ou très petite. Ces substances comprennent les acides formiques et aminoacétiques.
Dans les solvants non aqueux, les électrolytes inorganiques sont également capables d'augmenter la tension superficielle, et cela dépend du solvant.
Par exemple, lorsque l'iodure de sodium est introduit dans le méthanol, la tension superficielle augmente fortement ; pour l'éthanol, la tension superficielle est environ 2 fois supérieure. L'activité de surface des substances peut dépendre non seulement de la nature de la substance, mais également des propriétés du solvant. Si un solvant a une tension superficielle élevée, alors ce soluté peut présenter une activité de surface significative.
36. Théories de l'adsorption
Considérons les théories d'adsorption les plus courantes décrivant les différents types d'adsorption sur l'interface « solide-gaz » ou « solide-solution ».
La théorie de l'adsorption monomoléculaire par I. Langmuir.
1. L'adsorption est localisée et est causée par des forces proches des forces chimiques.
2. L'adsorption ne se produit que sur les centres actifs - saillies ou dépressions à la surface de l'adsorbant, caractérisées par la présence de valences libres. Les centres actifs sont considérés comme indépendants et identiques.
3. Chaque centre actif est capable d'interagir avec une seule molécule d'adsorbat ; une seule couche de molécules adsorbées peut se former à la surface.
4. Le processus d'adsorption est réversible et équilibré; la molécule adsorbée est retenue par le centre actif pendant un certain temps, après quoi elle est désorbée ; Après un certain temps, un équilibre dynamique s'établit.
La valeur d'adsorption maximale possible g o est atteint à condition que tous les centres actifs soient occupés par des molécules d'adsorbat. Équation d'isotherme d'adsorption monomoléculaire reliant la valeur d'adsorption g avec concentration en adsorbat DE, ressemble à:
où b est une valeur constante pour un couple « adsorbant-adsorbat » donné (rapport des constantes de vitesse de désorption et d'adsorption), numériquement égale à la concentration d'adsorbat à laquelle la moitié des centres actifs sont occupés.

Le graphique de l'isotherme d'adsorption de Langmuir est représenté sur la figure 2. La constante b on définit graphiquement en traçant une tangente à l'isotherme d'adsorption au point DE= 0. Lors de la description du processus d'adsorption des gaz dans l'équation, la concentration peut être remplacée par une valeur proportionnelle de la pression partielle. Théorie de l'adsorption monomoléculaire I.Langmuir applicable pour décrire les processus d'adsorption des gaz et des substances dissoutes à basse pression (concentrations) de l'adsorbat.
La théorie de Polanyi de l'adsorption polymoléculaire décrit des isothermes d'adsorption en forme de s, dont la forme indique l'interaction possible des molécules adsorbées avec l'adsorbat.
1. L'adsorption est causée par des forces physiques.
2. La surface de l'adsorbant est homogène, il n'y a pas de centres actifs ; les forces d'adsorption forment un champ de force continu près de la surface de l'adsorbant.
3. Les forces d'adsorption agissent à une distance supérieure à la taille de la molécule d'adsorbat, c'est-à-dire qu'il existe un certain volume d'adsorption près de la surface de l'adsorbant, qui est rempli de molécules d'adsorbat pendant l'adsorption.
4. L'attraction d'une molécule d'adsorbat par la surface de l'adsorbant ne dépend pas de la présence d'autres molécules dans le volume d'adsorption, ce qui permet une adsorption polymoléculaire.
5. Les forces d'adsorption ne dépendent pas de la température et, par conséquent, avec un changement de température, le volume d'adsorption ne change pas.
Équation de Freundlich. La surface de l'adsorbant est hétérogène, il y a une interaction entre les particules adsorbées, les centres actifs ne sont pas totalement indépendants les uns des autres. G. Freindlich ont suggéré que le nombre de moles de gaz ou de soluté adsorbé par unité de masse de l'adsorbant (l'adsorption dite spécifique X/m), doit être proportionnelle à la pression d'équilibre (pour le gaz) ou à la concentration d'équilibre (pour les substances adsorbées à partir de la solution) de l'adsorbant porté à une certaine puissance, qui est toujours inférieure à l'unité :
X / m = ap n X / m = ca n.m.
exposants n et facteur de proportionnalité un déterminée expérimentalement.
37. Thermodynamique du processus d'adsorption. Équation d'adsorption de Gibbs
Pour étudier le phénomène d'adsorption à la frontière "solution - gaz", il est nécessaire d'établir une relation entre l'excès de substance adsorbée dans la couche en surface ( g), concentration en tensioactif en solution ( Avec) et la tension superficielle ( σ ) à la limite de phase « solution-gaz ». Il est plus opportun de considérer les phénomènes du point de vue thermodynamique et de relier l'adsorption d'un soluté à une modification de l'énergie libre de la surface ou de sa tension superficielle. Ce lien a été établi W. Gibbs dans 1876, qui a été nommé "Équation d'adsorption de Gibbs":
g = – Avec / RT X dσ/cc.
Vous pouvez encore imaginer Équation de Gibbs, basé sur la thermodynamique, utilisant le potentiel isobare-isotherme g, potentiels chimiques µ 1 et µ 2 , et aussi en utilisant n 1 et n 2 le nombre de moles des composants. Après l'avoir analysé en tenant compte de l'entropie S, le volume V et la pression P, on peut écrire l'équation suivante :
dG=– TSD+VdP+σds+ μ 1 j n 1 + μ 2 jours 2.
Nous l'assimilons à zéro, et en tenant compte de la température et de la pression constantes, il se simplifie en une équation de la forme :
Dakota du Sud σ + n 1 j μ 1 + n2d μ 1 = 0.
En tenant compte du fait que pour les solutions diluées, le potentiel chimique du deuxième composant s'exprime comme suit :
μ 2 = μ 2 0 +RT dans c,
et étant donné que la température est constante
dμ 2 =RDTNC,
en remplaçant cette équation par
![]()
on obtient l'équation d'adsorption de Gibbs désirée. Sur la base de l'équation, on peut voir que si la tension superficielle σ augmente avec la concentration Avec, alors la concentration de la substance dissoute sur la couche superficielle est inférieure à celle du volume de la solution (appelée adsorption négative), et si la tension superficielle σ diminue avec l'augmentation de la concentration Avec, alors la concentration dans la couche est supérieure à celle dans le volume (adsorption positive), et, enfin, si σ ne dépend pas de Avec, alors la concentration de la substance dans la couche à la surface et dans le volume est la même. L'équation de Gibbs a été dérivée en utilisant la thermodynamique. Il est difficile de vérifier cette équation dans la pratique, ce qui est dû à la complexité de la détermination de la concentration d'une substance dissoute dans une surface stratifiée. Expérimenté B.McBen a constaté qu'une très fine couche de liquide était coupée de la surface de la solution à l'aide de l'appareil. Une détermination plus poussée de tous les paramètres de l'équation de Gibbs a montré que les valeurs d'adsorption trouvées expérimentalement coïncidaient avec les valeurs calculées à l'aide de l'équation de Gibbs dans l'erreur expérimentale. En raison de l'homogénéité et de la douceur de la surface de tout liquide, lors de l'étude de l'adsorption à sa surface, les idées habituelles sur les centres actifs sont totalement inapplicables. À la température critique, la différence entre les phases adjacentes disparaît et la tension superficielle, en règle générale, devient égale à zéro. L'adsorption des gaz et des vapeurs a une application pratique si large que dans la littérature, en particulier dans la technique, vous pouvez trouver ce concept, qui n'est utilisé qu'en relation avec les processus à la surface des solides.
Ce concept, ainsi que les schémas d'adsorption les plus généraux, comme l'équation de Gibbs considérée, est applicable à toutes les limites de phase. En utilisant l'équation de Gibbs et toutes les dispositions qui en découlent, après avoir déterminé la valeur de Г, il est possible de construire une isotherme d'adsorption.
38. Particularités de l'adsorption sur des matériaux microporeux. Théorie du potentiel de Polan. Potentiel d'adsorption
La théorie de Glade considère l'adsorption physique non localisée, qui est directement due aux forces de van der Waals entre l'adsorbant et l'adsorbat (cela peut être considéré comme la première position). La deuxième position de cette théorie est la notion de champ de force (ou de potentiel) de l'adsorbant, qui s'étend sur une distance considérable de la surface ; la couche d'adsorption qui apparaît dans ce champ est polymoléculaire. Si l'on considère l'adsorption des gaz, la densité de cette couche diminue le long d'une certaine normale à la surface. Si l'on considère l'adsorption de vapeur, une couche liquide d'une certaine épaisseur se forme à la surface. Le champ dans la théorie de Polanyi est considéré comme une série de surfaces équipotentielles, chaque surface correspond à une certaine valeur du potentiel ε , et chaque surface suivante sera plus petite que la précédente. Chacune de ces surfaces dans l'espace découpe des couches d'un certain volume, désignées par v je. La tâche de la théorie de Polanyi est de trouver la transition à partir des coordonnées habituelles de l'isotherme ( x, p) aux paramètres de champ ε je et v je, avec en outre l'établissement du lien entre ces principaux paramètres. La première partie du problème, qui a été posée par Polyani, est assez compliquée et, dans de nombreux cas, ne peut pas avoir de solutions définitives, mais pour le cas de l'adsorption de vapeur, cette partie du problème est résolue en première approximation très simplement. Pour une couche d'adsorption liquide, la partie remplie du volume sera égale à :
v je \u003d x (M / j),
où ré est la masse volumique de la substance à l'état liquide.
Dans sa théorie, M. Polyany introduit une autre disposition sur l'absence du soi-disant. dépistage sur le terrain dans le processus d'adsorption, la valeur ε dans cette théorie de l'espace est une valeur constante (quelque chose comme un potentiel gravitationnel) indépendamment du fait que certaines molécules d'adsorbat existent entre un point donné et une surface solide, ou que tout l'espace soit libre. Polyani présente le concept potentiel d'adsorption ε , qui est le travail isotherme de compression de la vapeur lorsqu'elle est transférée de la pression d'équilibre R dans la phase brute loin de la surface jusqu'à la région de la couche superficielle à pression de vapeur saturante p 0 alors l'expression pour déterminer le potentiel ressemblera à:
ε = RT dans R 0 / R.
A l'aide d'une telle équation, on peut passer des coordonnées x, p aux coordonnées ε et v et obtenir une courbe, que l'on appelle "caractéristique". Polanyi a découvert dans ses expériences que de telles courbes, construites à partir des données expérimentales des isothermes obtenues, ont la propriété suivante : elles sont invariantes par rapport à T, ou, en d'autres termes, toutes les courbes de ce type peuvent reposer sur une courbe ε −ε .
M. Polyany a accepté cette position comme postulat, c'est-à-dire :
Cette propriété de Polyani est d'une grande importance pratique, elle peut construire une famille d'isothermes à partir d'une isotherme d'adsorption expérimentale.
La théorie de Polanyi ne donne pas d'expression analytique pour l'isotherme ou la fonction du potentiel en fonction du volume, mais permet de calculer la coordonnée pour une température donnée si au moins une isotherme est connue. Ce résultat est très important pour les calculs technologiques, car pour des gaz similaires sur un même adsorbant, les courbes d'adsorption peuvent s'avérer proches les unes des autres et peuvent dans de nombreux cas se superposer.
39. Courbe caractéristique d'adsorption. Invariance en température et affinité des courbes caractéristiques
Le champ de force qui apparaît à la surface de l'adsorbant peut être similaire à bien des égards au champ gravitationnel. Dans le domaine de l'adsorption, on peut représenter des surfaces potentielles, c'est-à-dire des surfaces pour lesquelles un même potentiel d'adsorption est caractéristique. Sous le concept de potentiel d'adsorption θ doit être compris comme rien de plus que le travail effectué contre les forces d'adsorption lors du déplacement d'une mole d'adsorbat d'un certain point du champ vers une certaine phase gazeuse. Le potentiel d'adsorption maximal existera à la frontière « adsorbant - volume d'adsorption ». Mais à la frontière "volume - phase gazeuse" (c'est là que s'arrête l'action des forces d'adsorption), le potentiel d'adsorption doit être égal à zéro. L'évolution du potentiel d'adsorption avec une évolution du volume d'adsorption peut être représentée sous forme de courbes. Cela a d'abord été fait par M. Polyani. De tels types de courbes ne dépendent pas de la température et peuvent être caractéristiques de chaque adsorbant particulier ; ces types de courbes sont généralement appelés courbes caractéristiques d'adsorption. La théorie de l'adsorption polymoléculaire suppose que l'équation d'état du gaz s'applique à la quantité d'adsorption. Par conséquent, les isothermes qui caractérisent la dépendance de la densité de l'adsorbat au volume pour différentes températures ressemblent aux isothermes de la dépendance de la pression au volume. À basse température, les forces d'adsorption sur la surface peuvent provoquer la condensation de la vapeur en un liquide d'une certaine densité. À des températures inférieures à la température critique, lors de la condensation, tout le volume d'adsorption sera rempli de liquide. Dans ce cas, la courbe d'adsorption sera quasiment parallèle à l'axe des abscisses, ce qui est lié à la faible compressibilité du liquide. Ensuite, la courbe d'adsorption à la frontière "volume - phase gazeuse" chute brusquement et, par conséquent, la densité de l'adsorbat atteint la valeur d'une certaine densité de la phase gazeuse. À des températures supérieures à la critique, l'adsorbant peut se comporter comme un gaz parfait, et le graphique sera exprimé comme une isotherme de la dépendance pour un gaz parfait, à condition que pV = RT. Dans ces conditions, le gaz adsorbé aura un maximum de densité à la surface même de l'adsorbant et un minimum de densité au voisinage immédiat de la phase gazeuse. De plus, dans ce cas, il est important de noter que la densité de l'adsorbat dans la couche d'adsorption n'atteint nulle part la densité du liquide lui-même. Et si la température est très proche de la critique, la dépendance de la densité au volume sera exprimée par une courbe proche en apparence de l'isotherme, qui est décrite équation de van der Waals. Dans ce scénario, une partie de la substance adsorbée sera dans le volume adsorbé à l'état liquide, et une partie de la substance adsorbée sera à l'état gazeux. Ensuite, la courbe diminuera le plus fortement dans la section qui correspond au passage du liquide au gaz. Si une courbe caractéristique est construite à partir de l'isotherme d'adsorption expérimentale de l'un des adsorbants, et connaissant les coefficients d'affinité correspondants pour un autre adsorbant, on peut trouver l'isotherme d'adsorption et la construire pour un autre adsorbant. La théorie potentielle de l'adsorption permet de calculer différentes isothermes d'adsorption de différentes vapeurs sur le même adsorbant, de plus, selon la courbe caractéristique, qui est obtenue à partir de l'isotherme d'adsorption d'une vapeur, puisque le rapport du potentiel d'adsorption ne dépend pas sur les volumes d'adsorption.
affinité(du latin affinis - "apparenté") - chromatographie d'affinité. La méthode de purification et de séparation des protéines est basée sur leur interaction sélective avec un ligand lié de manière covalente à un support inerte (chromatographie d'affinité). La mesure de l'affinité d'un toxique pour un récepteur est en fait une étude expérimentale de la relation entre la quantité d'une substance ajoutée au milieu d'incubation et la quantité de complexe toxique-récepteur formé à la suite de l'interaction.
Adsorption(du latin ad - on, at et sorbeo - j'absorbe), un changement (généralement - une augmentation) de la concentration d'une substance près de l'interface ("absorption à la surface"). Cause adsorption- insaturation des liaisons intermoléculaires près de la surface, c'est-à-dire l'existence d'un champ de force d'adsorption. Un corps qui crée un tel champ est appelé un adsorbant, une substance dont les molécules peuvent être adsorbées est appelée un adsorbat et une substance déjà adsorbée est appelée un adsorbat. Processus inverse adsorption est appelée désorption.
La nature du champ d'adsorption est différente. Si l'adsorption est associée à des liaisons de van der Waals, alors adsorption appelé physique. S'il s'agit de liaisons de valence, c'est-à-dire adsorption passe avec la formation de composés chimiques de surface, puis adsorption appelé un produit chimique chimisorption. Caractéristiques importantes chimisorption montre : irréversibilité, effets thermiques élevés (centaines de kJ/mol), caractère activé. Il existe de nombreux types intermédiaires adsorption entre physique et chimique adsorption. Par exemple, adsorption causée par la formation de liaisons hydrogène. Il existe également différents types de physique adsorption. L'occurrence la plus courante des forces d'attraction intermoléculaires de dispersion, en raison du fait qu'elles sont approximativement constantes pour les adsorbants avec une surface de toute nature chimique (non spécifique adsorption). Physique adsorption peut être causée par des forces électrostatiques (interaction entre ions, dipôles ou quadripôles) ; où adsorption déterminée par la nature chimique des molécules de l'adsorbant (la nature dite spécifique adsorption). La géométrie de l'interface joue également un rôle important. si la surface est plane, alors c'est adsorption surface ouverte, dans le cas d'une surface légèrement ou fortement courbée - environ adsorption dans les pores de l'adsorbant.
En théorie adsorption Distinguer statique (le système adsorbant-adsorbat est en équilibre thermodynamique) et cinétique (il n'y a pas d'équilibre).
Statique d'adsorption
Thermodynamique de l'adsorption
.Fondamentaux de la thermodynamique adsorption ont été créés par J. Gibbs dans les années 70. 19ème siècle Selon Gibbs, dans un système biphasé à l'équilibre près de l'interface, il y a un certain changement dans les valeurs locales de toutes les propriétés extensives (à l'exception du volume). Cependant, les phases sont considérées comme homogènes jusqu'à une certaine surface géométrique les séparant. Par conséquent, la valeur de toute propriété extensive pour le système dans son ensemble n'est pas égale à la somme des valeurs de cette propriété dans les phases homogènes et . La différence est attribuée à la phase de surface bidimensionnelle associée à la surface de séparation. Car la phase de surface n'a pas d'épaisseur, alors V0=+ et =-, où V- le volume.
Les représentations présentées permettent de ramener l'équation thermodynamique fondamentale à la forme :
où G est l'énergie libre de Gibbs, S est l'entropie, est la tension interfaciale, s est la zone d'interface et n je- le potentiel chimique correspondant et le nombre de moles je-ce composant. L'indice indique la valeur de la propriété respective dans la phase de surface. La transformation de Legendre permet de modifier l'équation (1) pour des conditions isothermes :
La quantité s'appelle le Gibbs adsorption et est désigné par le symbole G, (exprimé en mol/cm 2 ). Pour un système à deux composants :
La position de la surface de séparation peut être choisie arbitrairement. En particulier, le choix de cette disposition peut satisfaire la condition à 1 =0. Une telle surface est dite équimoléculaire. Pour cela, la désignation Г 2 = Г 2 (1) est introduite. Cela implique l'équation principale d'adsorption de Gibbs :
Si l'adsorbant est totalement insoluble dans l'une des deux phases, =const, et le passage de l'équation (2) à l'équation (3) ne nécessite pas la condition Г 1 =0. Ainsi, les Gibbs adsorption est l'excès de ce composant dans un système diphasique réel par rapport à un tel système dans lequel les deux phases seraient strictement homogènes jusqu'à la surface de séparation. En plus des quantités excédentaires de Gibbs adsorption, joue un rôle important dans sa théorie adsorption, compris comme le contenu complet du composant je dans l'espace O, qui présente des forces d'adsorption. Dénotant le contenu complet à travers un et en supposant que la composante je totalement insoluble dans l'une des phases brutes, on a :
où c je-concentration je-ème composant dans la phase en vrac. Pour les petits s je:
Adsorption peut se produire à n'importe quelle interface entre deux phases quelconques, en particulier à l'interface fluide-fluide (liquide-gaz, liquide-liquide) ou solide-fluide (solide-gaz, solide-liquide). Dans les systèmes fluide-fluide, α peut être mesuré en tant que fonction et déterminé expérimentalement Г 2 (1) à l'aide de l'équation (3). Dans le second cas, pour déterminer G 2 (1) est mesuré par n'importe quelle méthode n je 0 , , et les concentrations du i-ème composant dans ces volumes. A partir de là, G est calculé je(une) . Cette méthode est dite volumétrique (ou volumétrique). Avec la méthode pondérale (gravimétrique), la quantité est directement déterminée je-ème composant sur l'interface.
Isotherme d'adsorption
.Dans un système d'adsorption à l'équilibre, les paramètres qui déterminent l'équilibre sont un je pressions partielles R(ou avec je) et la température J. Ils sont liés par l'équation dite thermique :
À adsorption adsorbant individuel ( je=1) cette équation prend la forme :
Trois cas particuliers de l'équation thermique (lorsque T, r ou un- constantes) jouent un rôle particulier dans la théorie adsorption:
un=- équation isotherme adsorption,
T=- équation isobare adsorption,
R-- équation isostère adsorption.
Le type spécifique de fonctions et est déterminé par les caractéristiques du système considéré. Si l'un d'eux, par exemple, est connu pour n'importe quelle valeur T= const, alors évidemment les deux autres deviennent également connus. Dans ce cas, il n'est pas nécessaire que la forme analytique des dépendances soit connue. Ils peuvent être donnés empiriquement comme un ensemble de valeurs un, r et J.
En théorie adsorption généralement la question porte sur la forme de la fonction un=(p)r, c'est-à-dire à propos de l'équation isotherme adsorption. Ce problème est lié aux effets thermiques accompagnant adsorption. Lors du calcul de la variation des valeurs des principales fonctions thermodynamiques dans le cas de la transition dn moles d'adsorbant de la phase brute à la phase superficielle dans un système d'équilibre à p = const, deux cas sont possibles : dans le premier cas, seule la transformation de l'adsorbat en adsorbat est prise en compte, puisque l'adsorbant à adsorption thermodynamiquement inchangé et son rôle est de servir de source du champ d'adsorption ; dans le second, le changement d'adsorbant est également pris en compte.
Puisque le système est en équilibre, les potentiels chimiques de l'adsorbat et de l'adsorbat sont les mêmes ; entropie d'adsorbat due à une diminution de la mobilité des molécules à adsorption inférieure à l'entropie de l'adsorbant. Ainsi, avec un adsorbant inerte, l'enthalpie est toujours négative, c'est-à-dire adsorption exothermique. La prise en compte de l'évolution de l'entropie de l'adsorbant peut modifier cette conclusion. Par exemple, lors de la sorption par des polymères de substances dans lesquelles le polymère gonfle, l'entropie de ces derniers (due à une augmentation de la mobilité des macromolécules) peut augmenter si fortement que adsorption devient endothermique. Dans ce qui suit, seul l'exothermique adsorption.
Distinguer la chaleur intégrale, différentielle, isostérique et moyenne adsorption. chaleur intégrale Qégale à la perte d'enthalpie (à V= const - énergie interne constante) lors du changement adsorption de un 1 avant de un 2(dans un cas particulier, il peut s'agir d'un 1 \u003d 0): Q \u003d - (H 2 - H 1). Cette valeur est généralement rapportée à la masse de l'adsorbant et est exprimée en J/kg.
Chaleur différentielle q(J / mol) est égal à la perte d'enthalpie dH quand ça change un sur le un. Elle s'exprime par l'attitude q = - (dH/da). Il est évident que
La chaleur isostérique q st est prise égale à :
où est la différence entre les volumes molaires de l'adsorbat et de l'adsorbat. On peut montrer que  pour un adsorbant de gaz parfait :
pour un adsorbant de gaz parfait :
Le sens de l'introduction qsi en ce que sa détermination ne nécessite pas de données calorimétriques (telles que Q et q) et il peut être calculé par l'équation (9) à partir des résultats de mesure adsorption. Nous introduisons également la chaleur moyenne Q(J/mol):
Avec la croissance un paramètre Q toujours croissante, une q peut diminuer, augmenter ou rester inchangé. Avec la croissance un avec une surface non uniforme adsorption se produit dans des zones de moins en moins actives, ce qui entraîne une diminution q. Cependant, dans ce cas, les distances moyennes entre les molécules adsorbées diminuent, ce qui entraîne une augmentation des forces d'attraction entre elles, et q augmente. Le rapport entre les deux effets mentionnés détermine l'évolution de la dépendance q=f(a). Au très grand un les forces répulsives commencent également à prédominer dans cette région. q diminue toujours avec la croissance un.
Pour de très petites couvertures de surface, l'équation isotherme adsorption a la forme de l'équation de Henry :
où K H est le coefficient de Henry. En effet, pour de très petites un La couche d'adsorption est similaire à un gaz parfait bidimensionnel, donc son équation d'état est : rt, où est la pression bidimensionnelle, est la surface occupée par une mole de la substance. Par conséquent, en tenant compte de =-, et en utilisant l'équation (3), nous obtenons l'équation (12). L'équation de Henry exige que qétait constant. Pour les gros remplissages, cette équation cesse de tenir. Par conséquent, G. Freindlich (1906) a proposé de décrire les isothermes adsorption l'équation empirique suivante (l'équation de Freundlich) :
où k et n- constantes. Cette équation est souvent utilisée comme formule d'interpolation, bien que pour les petits R n'entre pas dans l'équation (12), et à très grande échelle R conduit à une augmentation illimitée, ce qui est incompatible avec l'expérience un.
Théorie rigoureuse des isothermes adsorption a été créé par I. Langmuir (1914-18). La théorie est basée sur ce qui suit. modèle : 1) la surface de l'adsorbant est un ensemble de centres actifs énergétiquement identiques sur lesquels les molécules d'adsorbat sont adsorbées (localisées) ; 2) une seule molécule est adsorbée sur un centre ; à adsorption une seule adsorption se forme. couche (monocouche); 3) adsorption sur ce centre n'affecte pas adsorption sur d'autres centres, c'est-à-dire l'interaction. les molécules adsorbées peuvent être négligées.
Modèle de Langmuir appelé. monomoléculaire localisé adsorption sur une surface uniforme. équation isotherme adsorption correspondant à ce modèle, peut-être. obtenu avec l'aide de méthodes (moléculaire-cinétique, thermodynamique, statistique-thermodynamique). Ainsi, l'adsorption l'équilibre peut être exprimé comme suit. schème:
Sans molécule. Adsorption en gaz + adsorption. centre de phase complexe (centre occupé)
La concentration de molécules dans le gaz est proportionnelle à p, la concentration de libre. centre-valeur ( à), où et T - nombre total de centres, nombre de centres occupés, concentration d'adsorption. valeur-complexe adsorption La constante d'équilibre est donc : K p \u003d p (un t - un)/ adsorption De là, nous obtenons l'équation de Langmuir :
où b-t. appelé adsorption coefficient égal à K p -1. Dans la région des très basses pressions pb" 1 et a = (a m b)p, qui correspond à l'équation de Henry, dans laquelle KH= un m b. Dans le domaine des très hautes pressions Br 1 et aa t; où adsorption ne dépend plus de la pression. Constante d'équilibre b-1 est lié à la valeur standard du potentiel isobare de la réaction :
Le modèle de Langmuir exige que le diff. chaleur et entropie adsorption ne dépendait pas du degré de remplissage de la surface.
l'équation (14) est une expression stricte correspondant au modèle de Langmuir, mais elle est rarement justifiée en pratique, puisque le modèle lui-même est idéalisé adsorption Doctrine de adsorption des années 20 20ième siècle en moyens. degré a été construit sur la base de l'affaiblissement ou de l'élimination de l'une ou l'autre hypothèse de Langmuir adsorption
Langmuir a déjà proposé une manière de décrire adsorption sur une surface inhomogène (c'est-à-dire sous l'hypothèse que tous les centres ne sont pas identiques). En combinant des centres identiques en groupes et en supposant que l'équation (14) s'applique à chaque groupe, nous pouvons supposer que adsorption sur toute la surface s'exprime par la somme des termes de l'équation (14) :
En supposant que le nombre d'adsorption les centres peuvent être décrit par une fonction continue de la distribution des valeurs de libre. énergie, Ya.B. Zel'dovich a obtenu de la formule (16) pour la fonction exponentielle une équation du type (13).
adsorption sur des surfaces inhomogènes - un grand chapitre de théorie adsorption Son principal tâche-solution de l'équation intégrale :
où f(p) - soi-disant. empirique isotherme adsorption, -que ou une autre f-tion de la distribution du nombre de centres sur les valeurs de libre. énergie,( b, p)- isotherme locale adsorption, qui est généralement considéré comme l'isotherme de Langmuir adsorption
De nombreuses tentatives ont été faites pour rejeter la deuxième hypothèse de Langmuir. adsorption Dans cette voie, la théorie des polymoléculaires adsorption, proposé par S. Brunauer, P. Emmet et E. Teller (théorie BET). La théorie postule qu'à des températures inférieures à la température critique, chaque molécule adsorbée dans la première couche (la chaleur d'adsorption Qi,), est le centre des molécules qui forment la deuxième couche, et ainsi de suite. On suppose que la chaleur adsorption dans toutes les couches, sauf la première, est égale à la chaleur de condensation. Ce modèle conduit à l'équation :
où c = exp[(q 1 -)/RT]. l'équation (18) en coordonnées a, p/p s correspond à une courbe en S. En coordonnées p/p s ,
isotherme adsorption selon l'équation (18) doit être linéaire. La pente de cette droite (généralement de l'ordre de 0,05 p/p s 0,30) et le segment coupé par celle-ci sur l'axe des ordonnées donnent les valeurs resp. à et Avec. L'utilisation généralisée de la théorie BET est due au fait que ses auteurs, en fait, considérant adsorption non localisé, identifiez la constante à pas avec le nombre d'adsorbants discrets. centres, mais avec le nombre de molécules d'adsorbat dans la première couche à l'emballage le plus proche (à R= ps). Par conséquent, en introduisant l'idée de la surface occupée par une molécule dans cette couche, nous acceptons :
où s- surface adsorbée adsorption En règle générale, l'isotherme est mesurée pour cette adsorption azote et prenons celui pour sa molécule = 0,162 nm 2. Un calcul similaire couramment effectué s selon le modèle de Langmuir n'est pas correct, car cette méthode ne s'applique évidemment qu'aux non localisés adsorption
dans la théorie polymoléculaire adsorption une grande contribution a été apportée par J. de Boer, qui a montré expérimentalement que la dépendance du nombre moyen de couches (plus que la première) sur toutes les surfaces se rapproche chimiquement. la nature, de p/p s est exprimée par une courbe universelle (appelée courbe en T). Il permet également d'estimer les surfaces des adsorbants.
Des tentatives ont été faites pour prendre en compte dans le modèle de Langmuir également l'interaction. entre adsorbeurs. molécules. Ainsi, T. Hill et J. de Boer, estimant que l'équation d'état de l'adsorption. couche est un analogue bidimensionnel de l'équation de van der Waals, nous avons obtenu ce qui suit. équation isotherme adsorption:
où= un/un t, un et b constantes de l'équation de van der Waals adsorption R. Fowler et E. Guggenheim, en tenant compte de l'interaction. adsorbeur molécules, a dérivé l'équation :
où est une constante associée à l'interaction par paires des molécules.
Il existe un autre mécanisme conduisant à des adsorption adsorbants en dessous de leur seuil critique. température sur adsorbants poreux à des valeurs relativement élevées p/p s. C'est la condensation capillaire. Si un ménisque d'adsorbat concave se forme dans un pore, la condensation y commence à p/p s Selon l'équation de Kelvin :
où est la tension superficielle de l'adsorbat, V -le sien volume molaire, rayon r de courbure du ménisque adsorption La condensation capillaire entraîne une forte augmentation de l'isotherme adsorption Dans ce cas, le soi-disant est souvent (mais pas toujours) observé. adsorption hystérésis, c'est-à-dire décalage d'adsorption. et désorbe. branches de l'isotherme. En règle générale, cela est dû au fait que la forme du ménisque à adsorption et la désorption ne correspondent pas.
La condensation capillaire est utilisée pour déterminer la taille des pores de l'adsorbant adsorption Selon l'équation (22) pour chaque valeur p/p s calculer le rayon de courbure du ménisque adsorption De là, compte tenu de l'épaisseur de l'adsorption. couche (par exemple, le long de la courbe en t), la forme de la région de transition de la couche au ménisque et la dépendance à la courbure à très petit r , trouver la taille linéaire (rayon effectif r ef) des pores remplis à un p/p s. Le volume de ces pores est déterminé par la croissance adsorption en ce point sur l'isotherme. A partir des données obtenues, une courbe de distribution du volume des pores selon leurs rayons est construite. La méthode est applicable à r ef 1,5 nm. Habituellement, le calcul est effectué par désorption. branches de l'isotherme, mais plus strict moderne. la théorie exige que les deux branches soient prises en compte pour construire la courbe.
Théorie potentielle de l'adsorption et théorie du remplissage volumique des micropores. Modèle adsorption, fondamentalement différent de celui de Langmuir, a été proposé en 1914 par M. Polyaki. Selon ce modèle, il y a adsorption potentielle près de la surface de l'adsorbant. champ de force décroissant avec la distance à la surface. Il en résulte que la pression de l'adsorbant, qui est égale à p loin de la surface, augmente près de celle-ci et atteint à une certaine distance la valeur p s à laquelle l'adsorbant se condense. Le volume de la couche entre l'interface et le géome. lieu des points où p = p s , rempli de liquide, qui est attribué aux valeurs normales de physique. propriétés du liquide en vrac. Isotherme réversible travail e adsorption. forces, déterminées par l'équation = RTlnp / p s, appelées. adsorption potentiel, et tout le concept est une théorie potentielle adsorption Pour un volume donné V adsorption la couche est potentiellement dépendante de la température (en raison de l'indépendance des forces de dispersion par rapport à la température). Cette invariance de température permet de recalculer adsorption d'un t-ry à l'autre, bien que les équations isothermes adsorption il n'était pas possible de déduire sur la base de la théorie énoncée. Le modèle Polyani a été largement et avec succès utilisé par de nombreuses personnes. auteurs, cependant, il contenait deux dispositions très vulnérables: 1) l'hypothèse que l'adsorption la plus fine. le film a des valeurs physiques normales. propriétés du liquide en vrac (cette hypothèse n'a pas été confirmée par des expériences); 2) invariance en température de la fonction =f(V), sous-jacente à la théorie n'a été approximativement confirmée par l'expérience que pour des adsorbants très finement poreux.
Utilisant la théorie du potentiel, M.M. Dubinin a proposé et développé la théorie du remplissage volumétrique des micropores (TOZM). Il a été postulé que cette théorie ne s'applique qu'aux adsorbants microporeux. Une caractéristique de tels adsorbants, dans lesquels les dimensions linéaires des pores sont de r1 nm, est que tout le volume de leurs pores est « rempli » d'adsorbants. champ. Par conséquent, lorsque adsorption ils ne sont pas remplis en couches, mais volumétriquement. La valeur dans le cas considéré n'est pas l'adsorption. potentiel, et jusqu'au signe du produit chimique. potentiel d'adsorbat, mesuré à partir du niveau de produit chimique. potentiel d'un liquide normal à la même température. L'ensemble des pores adsorbants est divisé en trois classes : les micropores ( r 0,6 nm), mésopores (0,6 nm-20 nm) et macropores ( r 20 nm). adsorption dans les micropores se produit selon le schéma TOZM, c'est-à-dire volumétriquement, dans les mésopores - selon le mécanisme de remplissage couche par couche, complété par une condensation capillaire. Macropores lors de l'adsorption. l'équilibre ne joue aucun rôle.
Présentation du concept de distribution f-tsii des volumes de pores sur les valeurs de produits chimiques. potentiel d'adsorption en eux, M.M. Dubinin et L. V. Radushkevich ont obtenu l'équation de l'isotherme d'adsorption TOZM, qui est généralement écrite dans ce qui suit. formulaire:

où n, E et un 0 -paramètres ( un 0 = unà p = ps). Dépendance à la température un 0:
où= -(da 0 /dT); un 0 0 = un 0à T \u003d T 0. Choix P et E pratiquement indépendant de la température. Dans la plupart des cas P= 2. Uniquement pour les cas où les manches initiales adsorption très grand n > 2. Pour recalculer les isothermes adsorption d'un adsorbant à l'autre, on suppose approximativement que E 1 /E 2 P 1 /P= et que a 01 /a 02 V 1 /V 2 , où P je- parachor, Vi- volume molaire d'adsorbant adsorption
Chaque adsorbant microporeux est caractérisé selon le TOZM par deux paramètres : W- volume microporeux ( W 0 = = une 0 V 0) et E 0 -caractéristique. énergie; W0 et E 0 désignent l'adsorbant standard, généralement le benzène.
En utilisant la notion qu'il existe des pores de tailles différentes dans un adsorbant réel, et en introduisant la distribution des valeurs E s variance égale à F. Stekli a proposé une généralisation de l'équation (23), appelée équation de Dubinin-Stöckli :

où B0- constante associée à E dans l'équation (23), et y= ![]() . Car dans l'absorption la technique du naib. ce sont les adsorbants microporeux (charbons actifs, zéolithes, xérogels finement poreux) qui se sont généralisés, le TOZM n'est pas utilisé qu'en physique et en chimie. recherche, mais aussi dans les calculs d'ingénierie.
. Car dans l'absorption la technique du naib. ce sont les adsorbants microporeux (charbons actifs, zéolithes, xérogels finement poreux) qui se sont généralisés, le TOZM n'est pas utilisé qu'en physique et en chimie. recherche, mais aussi dans les calculs d'ingénierie.
Adsorption de mélanges gazeux et liquides. En pratique, il s'agit toujours non pas d'un adsorbant individuel, mais d'un mélange de gaz ou de solutions liquides. Une généralisation de la théorie s'impose donc adsorption pour le cas d'un adsorbant multicomposant adsorption En principe, on peut partir de n'importe quel modèle adsorption et l'étendre à ce cas. À adsorption mélange de gaz, ceci est réalisé non seulement par une plus grande complication des équations, mais aussi en y introduisant des additions. empirique paramètres associés à ou à l'interaction. molécules hétérogènes ou, plus généralement, avec l'influence de certains in-in sur le coefficient. les activités des autres. Seul le modèle de Langmuir permet d'obtenir l'équation isotherme adsorption mélanges sans paramètres non inclus dans les équations pour adsorption in-in individuel. Pour ce faire, il suffit de prendre en compte que lors de l'adsorption du ke composant d'un mélange je composants faisant partie de l'adsorption. les centres peuvent être occupée par d'autres molécules. C'est pourquoi:
Lorsque adsorption solutions liquides, quelle que soit leur concentration, toute la surface de l'adsorbant est remplie de adsorption Ainsi adsorption molécules du k-ème composant s'accompagne du déplacement d'un certain nombre de molécules des composants restants, c'est-à-dire adsorption est compétitif.
Distinguer moléculaire et ionique adsorption solutions. Le premier se produit lorsque adsorption solutions de non-électrolytes, la deuxième solution d'électrolytes. Moléculaire adsorption, en règle générale, est exprimé sous forme de valeurs redondantes. Caractère compétitif adsorption provoque la valeur un mb à la fois positif et négatif. exprimer adsorption je-de ce composant en f-tion de sa fraction molaire dans la solution x je-, nous avons ce G je= O en x je= 0 et x je= 1 (une éventuelle modification du volume de la substance dans la couche d'adsorption est négligée). Par conséquent, l'isotherme adsorption a un ou plusieurs extrêmes.
équation isotherme adsorption les solutions binaires de non-électrolytes, corroborées de manière fiable thermodynamiquement, ont la forme :

où l'indice s indique l'adsorption. phase, - ( dn s 2 /dn s 1) montre combien de moles du deuxième composant sont déplacées par une mole du premier, la différence entre les termes (parties standard) du produit chimique. potentiel qui ne dépend que de la température.
Principal le problème de l'utilisation de cette équation et d'un certain nombre d'autres équations isothermes adsorption-détermination de la dépendance du coefficient. l'activité des composants dans l'adsorption. couche de sa composition adsorption La question la plus importante dans l'application adsorption pour la séparation ou la purification de substances - sélection d'un adsorbant sélectif par rapport à cette solution de composant adsorption
Ionique adsorption, en règle générale, n'est pas équivalent adsorption Les préim sont adsorbés en surface à partir de la solution d'électrolyte. cations ou anions. Grâce à l'électrique (Coulomb) forces sur la surface se forment double couche électrique.
Si l'adsorbant contient des ions ou des fonctions de surface. groupements capables de s'ioniser dans un solvant donné, il se produit alors un échange d'ions entre l'adsorbant et la solution d'électrolyte. L'adsorbant dans ce cas est appelé. échangeur d'ions.
Cinétique d'adsorption
adsorption, comme tout processus réel, se produit dans le temps. Donc la théorie complète adsorption devrait contenir une section sur la cinétique adsorption acte élémentaire adsorption presque instantanément (à l'exception de la chimisorption). Donc les dépendances temporelles adsorption sont définis pour l'essentiel par le mécanisme de diffusion, c'est-à-dire l'apport de l'adsorbant à l'endroit adsorption Si un adsorption sur la surface ouverte n'est pas instantanée, un tel processus se produit dans la région de diffusion externe ; alors que les lois de la diffusion ne sont pas spécifiques à adsorption Dans le cas des adsorbants poreux, en plus de ext. diffusion, un rôle important commence à jouer vnutr. diffusion, c'est-à-dire transfert de l'adsorbant dans les pores de l'adsorbant en présence d'un gradient de concentration dans ceux-ci. Le mécanisme d'un tel transfert peut dépendre de la concentration de l'adsorbat et de la taille des pores.
Il existe une diffusion moléculaire, de Knudsen et de surface (Volmer). La diffusion moléculaire s'effectue si la longueur est libre. la gamme de molécules dans les pores est inférieure à la taille des pores, la longueur de Knudsen est si cette longueur dépasse la taille des pores. Lors de la diffusion en surface, les molécules se déplacent sur la surface de l'adsorbant sans transition vers la phase brute. Cependant, les valeurs du coefficient les diffusions ne sont pas les mêmes pour les différents mécanismes de diffusion. Dans de nombreux cas, il n'est pas possible d'établir expérimentalement exactement comment se produit la diffusion, et donc la soi-disant. coefficient effectif. diffusion, décrivant le processus dans son ensemble.
Principal expérimental matériel sur la cinétique adsorption sert le soi-disant. cinétique courbe, c'est-à-dire f-tion \u003d a / a égal \u003d f(t) où est relatif adsorptionégal au rapport de la valeur actuelle de l'adsorption unà unégale à sa valeur au moment t. Interpréter la cinétique courbe dans le cas le plus simple, on suppose que le grain d'adsorbant a une structure poreuse parfaitement homogène en volume (ce modèle est dit quasi-homogène). moyens. amélioration du modèle quasi-homogène - la notion que chaque grain contient des régions avec des pores plus grands et plus fins. La diffusion dans un tel grain est décrite par deux déc. coefficients.
Dans le cas d'une surface ouverte, en prenant le modèle de Langmuir, il est facile d'obtenir la cinétique. l'équation adsorption Le taux d'approche de l'équilibre est la différence des vitesses adsorption et désorption. En supposant, comme d'habitude en cinétique, que les vitesses des processus sont proportionnelles aux concentrations des substances en réaction, nous avons :
où k ads et k dec sont respectivement les constantes de vitesse. adsorption et désorption. La pression dans la phase gazeuse est supposée constante. Lors de l'intégration de cette équation de t= 0 à n'importe quelle valeur t on a:
Donc, pour f on a := égal. Nous avons donc finalement :
où k = k annonces + k déc.
Effet de la température sur la vitesse adsorption exprimée par une équation similaire à l'équation d'Arrhenius adsorption Avec l'augmentation de la température, k ads augmente de façon exponentielle. Car la diffusion dans les pores de l'adsorbant est associée à la maîtrise de l'activation. barrières, les dépendances en température de k ads et k des ne sont pas les mêmes.
La connaissance des taux de diffusion est importante non seulement pour la théorie adsorption, mais aussi pour le calcul de prom. adsorption processus. Dans ce cas, ils ne traitent généralement pas des grains individuels de l'adsorbant, mais de leurs couches. La cinétique du processus dans la couche se traduit par des dépendances très complexes. En chaque point de la couche à un instant donné, la valeur adsorption est déterminé non seulement par la forme de l'équation isotherme adsorption et les lois de la cinétique du processus, mais aussi aéro- ou hydrodynamiques. conditions d'écoulement du gaz ou du liquide autour des grains. La cinétique du processus dans la couche adsorbante, contrairement à la cinétique dans un seul grain, est appelée. dynamique adsorption, dont le schéma général de résolution des problèmes est le suivant: un système de différentiels est compilé. équations aux dérivées partielles, tenant compte des caractéristiques de la couche, de l'isotherme adsorption, caractéristiques de diffusion (coefficient de diffusion, types de transfert de masse le long de la couche et à l'intérieur des grains), aéro- et hydrodynamiques. fonctionnalités de flux adsorption Les conditions initiales et aux limites sont définies. La solution de ce système d'équations conduit en principe aux valeurs des grandeurs adsorptionà un instant donné en un point donné de la couche. En règle générale, l'analyse la solution ne peut être obtenue que pour les cas les plus simples, par conséquent, un tel problème est résolu numériquement à l'aide d'un ordinateur.
Dans une étude expérimentale de la dynamique adsorption on fait passer à travers la couche adsorbante un courant gazeux ou liquide aux caractéristiques spécifiées et on examine la composition du courant sortant en fonction du temps. L'apparition de la substance absorbée derrière la couche appelée. percée, et le temps de percée - le temps de l'action protectrice. La dépendance de la concentration de ce composant derrière la couche sur le temps appelé. courbe de sortie. Ces courbes servent de principal expérimental matériel qui permet de juger les modèles de dynamique adsorption
Conception matérielle des procédés d'adsorption
Il existe de nombreuses technologies. technique d'adsorption. processus. Cyclique généralisé. (périodique-dich.) installations à lit fixe d'adsorbant, osn. dont le nœud est un ou plusieurs. adsorbeurs réalisés sous forme de colonnes creuses remplies d'adsorbant granulaire. Un flux gazeux (ou liquide) contenant des composants adsorbés traverse le lit d'adsorbant jusqu'à ce qu'il perce adsorption Après cela, l'adsorbant dans l'adsorbeur est régénéré et le flux de gaz est envoyé vers un autre adsorbeur. La régénération de l'adsorbant comprend plusieurs étapes dont la principale est la désorption, c'est-à-dire la désorption. libération de matière précédemment absorbée par l'adsorbant adsorption La désorption est effectuée par chauffage, dépressurisation de la phase gazeuse, déplacement (par exemple, vapeur vive) ou une combinaison de ces méthodes. Parce que les temps adsorption et régénération ne coïncident pas, sélectionner un nombre d'adsorbeurs fonctionnant simultanément et régénérés tel que l'ensemble du processus se déroule en continu.
Selon la technique. et économique considérations la régénération n'est pas menée à son terme adsorption Par conséquent, la capacité de travail de l'adsorbant est égale à la différence entre le maximum réalisable dans des conditions données adsorption et la quantité d'adsorbat restant dans l'adsorbant après régénération. Par conséquent, les isothermes adsorption correspondant au processus dans l'adsorbeur ne doit pas être trop raide.
Dans le schéma décrit, deux options sont possibles : 1) le produit cible est presque complètement adsorbé du flux gazeux, puis il est contenu dans le désorbat, d'où il est extrait d'une manière ou d'une autre ; 2) le produit cible est moins adsorbé que les autres composants du mélange gazeux, puis il est contenu dans le flux gazeux sortant. Selon la première option, par exemple, les unités de récupération des usines de viscose fonctionnent, capturant les gaz d'échappement et renvoyant le CS 2 au cycle. La productivité de telles installations atteint des centaines de milliers de m 3 de gaz épuré par heure ; charbon actif adsorbant avec des micropores pas trop fins, c'est-à-dire charbon, dans lequel la constante E selon TOZM (voir ci-dessus) est de 20-25 kJ / mol. Cette valeur E 0 correspond à une isotherme pas trop raide, qui offre de bonnes conditions de régénération. Ces charbons sont appelés récupération. La désorption est réalisée à la vapeur vive. Pour économiser de l'énergie, les flux de gaz froid et chaud passent par des échangeurs de chaleur.
Il est très important de sécher les gaz et les liquides, tels que les gaz de pétrole avant leur traitement ou leur nature. gaz avant transport; adsorbants de gel de silice ou zéolithes. La désorption est réalisée par chauffage. La désorption de la zéolithe étant associée à des coûts énergétiques élevés, on utilise un adsorbant combiné : principal. la masse d'humidité est absorbée par le gel de silice facilement régénéré et le post-séchage en profondeur par la zéolite.
Lors de la régénération thermique, le cycle complet comprend adsorption, chauffage de l'adsorbant, sa désorption et son refroidissement. Un grand nombre d'étapes détermine la faible intensité et la haute intensité énergétique du processus. adsorption Par conséquent, le soi-disant. installations à cycle court, dont le cycle complet en prend plusieurs. minutes. Dans ceux-ci, le gaz est fourni à l'adsorbeur sous la moyenne. pression, qui est ensuite relâchée et la désorption se produit. L'ensemble du processus est presque isotherme (l'écart par rapport à l'isothermie n'est causé que par le dégagement de chaleur adsorption et absorption de chaleur lors de la désorption). Étapes du cycle : adsorption, dépressurisation, désorption, montée en pression. Un exemple est une usine avec de la zéolithe pour produire de l'air enrichi en oxygène.
Dans les installations à couche mobile d'adsorbant (dans les soi-disant hypersorbeurs), ce dernier descend lentement sous l'influence de la gravité, est retiré du fond. parties de l'adsorbeur et entre dans le soi-disant. airlift, qui est un tuyau vertical parallèle à l'adsorption. colonne. Un flux d'air circule dans cette canalisation de bas en haut, ce qui remonte les grains d'adsorbant vers le haut. partie de la colonne. Le flux de gaz traité pénètre dans la partie médiane de l'adsorbeur et monte à contre-courant de l'adsorbant. Au sommet de la colonne, il y a un continu adsorption, en bas - régénération de l'adsorbant (voir aussi nettoyage par adsorption).
Dans les installations à lit d'adsorbant fluidisé ("bouillant"), le flux de gaz entrant dans l'adsorbeur par le bas met l'adsorbant en suspension. Cela augmente fortement l'efficacité du transfert de masse entre l'adsorbant et le gaz et réduit la durée adsorption et désorption. De telles installations ont une productivité élevée. Leur large diffusion est entravée par des besoins élevés en fourrure. la résistance des grains d'adsorbant (une résistance insuffisante entraîne des pertes importantes d'adsorbant du fait de son abrasion et de son entraînement hors de l'appareil).
Principal exigences pour les adsorbants : grand adsorbant. capacité, c'est-à-dire ils devraient être des corps dispersés avec un grand battement. surface ou avec un grand volume de pores; chim. la nature de la surface doit fournir une adsorption données in-in dans ces conditions ; chim. et thermique. durabilité, régénérabilité, disponibilité. max. les charbons actifs, les xérogels de certains oxydes (gels de silice, gels d'alumine...), les zéolithes se sont généralisés ; à partir d'adsorbants non poreux-tech. carbone (suie) et SiO 2 hautement dispersé (aérosil, « suie blanche »).
Domaines d'application de la technologie d'adsorption
Sur le phénomène adsorption fondée par de nombreux moyens de nettoyer l'air des impuretés nocives (voir. épuration des gaz), l'eau (voir Traitement de l'eau), ainsi que des sirops de sucre pour la fabrication du sucre, des jus de fruits et d'autres liquides dans les aliments. prom-sti, déchets d'huiles lubrifiantes. L'élimination de l'humidité en tant qu'impureté nocive des gaz et des liquides à l'aide d'adsorbants solides est l'une des branches importantes de l'adsorption. techniques (voir aussi séchage au gaz).
A l'adsorption. les procédés sont basés sur la séparation fine des mélanges de substances et l'isolement de certains composants à partir de mélanges complexes. Des exemples sont la séparation d'isomères d'alcanes afin d'obtenir des hydrocarbures normaux pour la production de tensioactifs, la séparation d'huiles dans la production de carburants. Pour les mélanges gazeux d'adsorption. les méthodes de séparation sont utilisées pour obtenir de l'air enrichi en oxygène (jusqu'à O 2 presque pur) ; dans de nombreux cas, ces méthodes concurrencent avec succès la distillation (voir. séparation d'air).
Le domaine d'application en plein essor des adsorbants. technologie médicale, où elle sert à extraire les substances nocives du sang (méthode d'hémosorption), etc. fiziol. liquides. Les exigences élevées en matière de stérilité posent une tâche très difficile de sélection des adsorbants appropriés. Ceux-ci comprennent des charbons actifs spécialement préparés.
Litt. : Brunauer S., Adsorption des gaz et des vapeurs, trad. de l'anglais, volume 1, M., 1948 ; de Boer Ya, La nature dynamique de l'adsorption, trad. de l'anglais, M., 1962; Adsorption et porosité, éd. M.M. Dubinina [et al.], M., 1976 ; Keliev N.V., Fundamentals of adsorption technology, 2e éd., M., 1984 ; Young D.M., Crowell A.D., Adsorption physique des gaz, L., 1962. MM Dubinin, V.V. Serpinski.
Choisissez la première lettre du titre de l'article :
Dans le cas de l'interaction de deux atomes :
U est l'énergie d'interaction ;
U = U + U TIRER.
 - Équation de Lennard-Jones
, c, b, m = const
- Équation de Lennard-Jones
, c, b, m = const
En cas d'interaction d'atomes avec une surface solide, il est nécessaire de résumer toutes les interactions.
x est la distance à la surface
r - le rayon d'action des forces d'attraction
dV - volume
n est le nombre de molécules de surface
ANNONCES U. est l'énergie d'interaction d'adsorption



Dans le cas de l'adsorption, l'attraction est renforcée. Et dans le cas d'interaction de type apolaire-apolaire, l'adsorption est majoritairement localisée dans les dépressions.
interaction électrostatique.
Adsorbant polaire - adsorbat non polaire
Adsorbant apolaire - adsorbat polaire
Un adsorbant polaire est un adsorbat polaire.
M  la molécule d'adsorbat est représentée comme un dipôle, et l'adsorbant est représenté comme un conducteur dans lequel la molécule d'adsorbat induit un dipôle en symétrie miroir par rapport à celui donné.
la molécule d'adsorbat est représentée comme un dipôle, et l'adsorbant est représenté comme un conducteur dans lequel la molécule d'adsorbat induit un dipôle en symétrie miroir par rapport à celui donné.
X - distance au milieu
Lors de l'interaction, le potentiel surgit:
 ,
,
 est le moment dipolaire.
est le moment dipolaire.
Le potentiel tend à prendre la valeur maximale, c'est-à-dire les dipôles ont tendance à s'orienter perpendiculairement à la surface.
Comme une augmentation de la température favorise la croissance du mouvement brownien, elle conduit à une décélération du processus d'adsorption.
Dans le cas de l'interaction électrostatique, l'adsorbat est majoritairement localisé sur les protubérances.
Équation fondamentale d'adsorption.
Dans le cas de l'adsorption, le composant est redistribué, ce qui signifie que le potentiel chimique change. Le processus d'adsorption peut être considéré comme la transition de l'énergie de surface en énergie chimique.
Volume de couche = 0, puis l'équation généralisée I et II de la loi de la thermodynamique :
T = const ; (1) = (2) => 

Pour un système à deux composants :
,
 ,
,
 =>
=>
=>
 - Équation d'adsorption de Gibbs
.
- Équation d'adsorption de Gibbs
.
Pour le cas d'adsorption de TV. corps - gaz :, 
 ,
,

 - isotherme
- isotherme
 - isobare
- isobare
 - isopycne
- isopycne
 - isostère
- isostère
Isotherme, isopycne, isostère sont liés les uns aux autres.
Car fonction d'adsorption 



isotherme de Henry isotherme de Langmuir



Thermodynamique. Adsorption.
Pour les médias condensés :
 ,
,
 ,
,
 - changement intégral de l'énergie de Gibbs
.
- changement intégral de l'énergie de Gibbs
.

P-pression sur une surface courbe, P S-pression sur une surface plane
 - potentiel d'adsorption
- potentiel d'adsorption
Changement différentiel d'entrapie 
 , Ã = const
, Ã = const
- changement d'entropie différentielle
- enthalpie différentielle d'adsorption
 - chaleur d'adsorption isostérique
- chaleur d'adsorption isostérique
 - chaleur de condensation
- chaleur de condensation
 - chaleur nette d'adsorption
- chaleur nette d'adsorption

 ,
,




Qa est la chaleur intégrale d'adsorption, 
Qra est la chaleur nette intégrale d'adsorption,

L'équation d'Henry
L'étude de l'adsorption est gênée par l'inhomogénéité de la surface, de sorte que les régularités les plus simples sont obtenues pour des surfaces homogènes.
Considérons l'interaction des gaz avec une surface solide, lorsqu'un gaz passe d'un état d'équilibre dans le volume à un état d'équilibre sur la surface. Ce cas est analogue à l'équilibre des gaz dans un champ gravitationnel.
 ,
,
 ,
=>
,
=> -L'équation d'Henry
-L'équation d'Henry
 - coefficient de répartition
- coefficient de répartition
Au cours du processus d'adsorption, une modification des potentiels chimiques se produit.
Pour la phase en vrac : 
Pour le gaz de surface : 
Dans un état d'équilibre  , c'est à dire.
, c'est à dire.

Dans l'équation de Henry, la constante ne dépend pas de la concentration
L'équation de Henry est valable dans le domaine des basses pressions et concentrations. Lorsque la concentration augmente, 2 types d'écarts à la loi d'Henry sont possibles :
1 - écarts positifs, D diminue, A diminue
2 - écarts négatifs, D - augmente, A - augmente.
Le type d'écart est déterminé par la prédominance de l'un ou l'autre type d'interaction adsorbant-adsorbat.
Avec une forte interaction adhésive, les coefficients d'activité augmentent - un écart positif. Dans le cas d'interactions cohésives, des déviations négatives sont observées.
adsorption monomoléculaire.
Isotherme de Langmuir.
Les régularités les plus simples ont été obtenues dans la théorie de Henry. Langmuir a proposé une théorie selon laquelle l'adsorption est considérée comme une réaction quasi-chimique. Où:
La surface est énergétiquement uniforme.
L'adsorption est localisée, chaque centre d'adsorption interagit avec une molécule d'adsorbat.
Les molécules d'adsorbat n'interagissent pas entre elles.
L'adsorption est monocouche.

 - surface,
- surface,  - adsorber,
- adsorber,  - complexe d'adsorption.
- complexe d'adsorption.
 , alors la concentration des sites d'adsorption :
, alors la concentration des sites d'adsorption :  ,
, - limiter l'adsorption.
- limiter l'adsorption.
 , alors la constante de réaction :
, alors la constante de réaction : 

 - Équation de Langmuir.
- Équation de Langmuir.
Adsorption versus concentration
1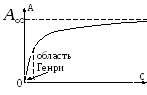 )
)
 ,
,
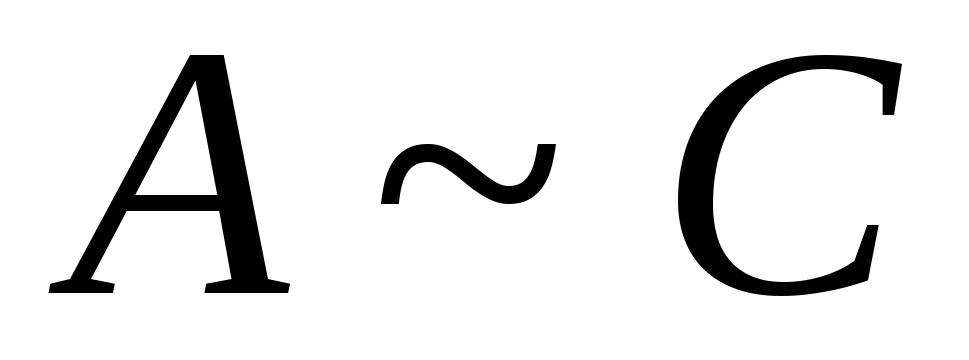
2) zone de fortes concentrations
 - limitation de l'adsorption, formation d'une couche monomoléculaire
- limitation de l'adsorption, formation d'une couche monomoléculaire
Pour l'énergie de Gibbs : .
g est le facteur d'entropie.
Dans le cas de l'isotherme de Henry, l'énergie de Gibbs caractérise le passage de l'adsorbat de l'état standard dans la masse à l'état standard en surface. Dans le cas de l'isotherme de Langmuir  caractérise le degré d'affinité de l'adsorbant et de l'adsorbat.
caractérise le degré d'affinité de l'adsorbant et de l'adsorbat.
 trouvé à partir de l'isobar de van't Hoff.
trouvé à partir de l'isobar de van't Hoff.

 , alors
, alors  , Par conséquent
, Par conséquent  .
.

 - degré de remplissage de la surface.
- degré de remplissage de la surface.




 - le nombre de postes vacants,
- le nombre de postes vacants,  - le nombre de places occupées.
- le nombre de places occupées.
 ,
,

Ceux. dans le domaine des fortes concentrations, le nombre de sites libres est inversement proportionnel à la quantité d'adsorbat.
Adsorption d'un mélange de gaz sur une surface homogène.
Dans ce cas, le processus d'adsorption est considéré comme deux réactions parallèles.
 (1)
(1)

 (2)
(2)


Adsorption d'un mélange de gaz sur une surface inhomogène.
Dans le cas d'une surface non homogène, il ne faut pas se limiter à des remplissages moyens.
Du fait de la concurrence, la localisation de divers adsorbats est possible dans différents types de sites.
Dans ce cas, la relation  .
.
 ,
,
 est la pression de vapeur saturante de l'adsorbat.
est la pression de vapeur saturante de l'adsorbat.
 ,
,
 est la chaleur d'adsorption.
est la chaleur d'adsorption.
"+" - dépendance symbatique, "-" - dépendance antibatique, "H" - pas de corrélation.
"+" - l'adsorption se déroule selon le même mécanisme. Dans les zones les plus énergétiquement favorables, un gaz ayant une forte affinité pour la surface est majoritairement adsorbé.
"-" - l'adsorption passe par divers mécanismes et jusqu'à un certain moment, il n'y a pas de compétition pour la surface.
L'adsorption monomoléculaire est principalement réalisée lors de l'adsorption physique de gaz à faibles valeurs p, ainsi qu'à l'interface liquide/gaz.
Adsorption polymoléculaire.
Théorie BET(Brunauer, Emmet, Teller).
Dans le cas où la formation d'une monocouche est insuffisante pour compenser l'énergie de surface, l'adsorption est polymoléculaire et peut être considérée comme le résultat d'une condensation forcée sous l'action des forces de surface.
Dispositions de base :
Lorsqu'une molécule d'adsorbat atteint le site occupé, un ensemble multiple se forme.
Alors que tu te rapproches pà p s le nombre de sites d'adsorption libres diminue. Au départ, le nombre de places occupées par les simples, les doubles, etc. augmente puis diminue. trousses.
À p =p s l'adsorption se transforme en condensation.
Il n'y a pas d'interactions horizontales.
Pour la première couche, l'isotherme de Langmuir est réalisée.
La surface est considérée comme un ensemble de sites d'adsorption. La condition d'équilibre dynamique est valide : le taux de condensation dans les lieux libres est égal au taux d'évaporation des lieux occupés.
a est le coefficient de condensation (la fraction de molécules condensées sur la surface) ;
 ,
,
Zm est le nombre maximum de places libres.
 - fréquence des vibrations des atomes dans la direction perpendiculaire à la surface.
- fréquence des vibrations des atomes dans la direction perpendiculaire à la surface.
Pour la première couche, les conditions d'équilibre dynamique sont :



 , alors
, alors
 - Équation de Langmuir.
- Équation de Langmuir.
Pour la deuxième couche sera vrai: 
Pour la ième couche : 
Pour simplifier, on suppose que a et ν sont les mêmes pour toutes les couches sauf la première. Pour toutes les couches sauf la première, la chaleur d'adsorption est constante. Pour la dernière couche, la chaleur d'adsorption est égale à la chaleur de condensation. En conséquence, l'équation
 (*)
(*)
C- constant,
Dans le cas de la théorie BET, la constante DE caractérise l'énergie de Gibbs d'adsorption pure. L'équation ne contient qu'une seule constante, et cette équation est également très importante pour déterminer la surface spécifique de l'adsorbant.
Étant donné que la chaleur est libérée par adsorption, la détermination des surfaces spécifiques est effectuée à basse température.
 ????????????
????????????


Le principal défaut de la théorie– Négligence des interactions horizontales au profit des verticales.
L'équation est dans la plage  de 0,05 à 0,3.
de 0,05 à 0,3.
Où  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 - l'interaction de l'adsorbat - adsorbat affecte.
> 0,3 - l'interaction de l'adsorbat - adsorbat affecte.
Prise en compte des interactions adsorbate-adsorbate.
Des interactions apparaissent lors de l'adsorption sur une surface non polaire de molécules ou de molécules ramifiées. capables de former des associations. Dans ce cas, la forme des isothermes d'adsorption change.
MAIS  le sorbant n'est pas polaire.
le sorbant n'est pas polaire.
Le graphique 1 correspond aux interactions adsorbat-adsorbat faible, adsorbat-adsorbant fort.
Le graphe 2 correspond à une forte interaction adsorbat-adsorbat, une forte interaction adsorbat-adsorbant.
Le graphique 3 correspond à une forte interaction adsorbate-adsorbat, une faible interaction adsorbate-adsorbant.
 ,
,

Dans le cas d'interaction entre molécules d'adsorbat, il faut tenir compte de l'évolution des coefficients d'activité. Et cette équation s'écrit :
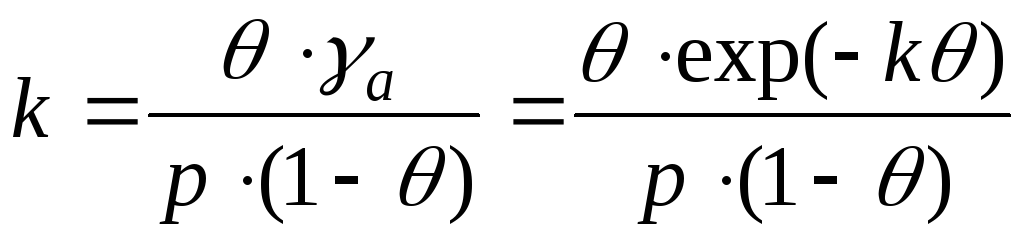 - l'équation de Frunkin, Fowler, Guggenheim.
- l'équation de Frunkin, Fowler, Guggenheim.
k est la constante d'attraction.
Théorie du potentiel de Polan.
Cette théorie ne dérive aucun type d'isotherme d'adsorption, mais permet de calculer des isothermes à une température différente.
Adsorption est le résultat de l'attraction de l'adsorbat à la surface de l'adsorbant due à l'action du potentiel d'adsorption, qui ne dépend pas de la présence d'autres molécules et dépend de la distance entre la surface et la molécule d'adsorbat.
 ,
,
 - potentiel d'adsorption.
- potentiel d'adsorption.
La surface étant inhomogène, la distance est remplacée par le volume d'adsorption  .volume d'adsorption est le volume compris entre la surface et le point correspondant à la valeur donnée
.volume d'adsorption est le volume compris entre la surface et le point correspondant à la valeur donnée  .
.
Potentiel d'adsorption est le travail de transfert de 1 mol de l'adsorbat hors du volume d'adsorption donné vers un point donné du volume d'adsorption (ou le travail de transfert de 1 mol de vapeur saturée de l'adsorbat, qui est en équilibre avec l'adsorbat liquide en l'absence de l'adsorbant, dans la phase vapeur en équilibre avec l'adsorbant).

Courbe caractéristique
 - potentiel d'adsorption,
- potentiel d'adsorption, 
Pour un adsorbant donné et différents adsorbats, on a : 
Pour différents types d'adsorbats  ,
,
où  potentiels pour les isothermes d'adsorption à des pressions relatives
potentiels pour les isothermes d'adsorption à des pressions relatives  pour l'adsorbat 1 et pour l'adsorbat 2. Ce rapport est une valeur constante.
pour l'adsorbat 1 et pour l'adsorbat 2. Ce rapport est une valeur constante.
 - coefficient d'affinité
- coefficient d'affinité
Théorie de la condensation capillaire.
Le déroulement du processus d'adsorption dépend largement de la structure du corps poreux.
|
microporeux | |
|
Poreux de transition | |
|
Macroporeux |
Dans le cas des sorbants microporeux, les champs de forces d'adsorption se recouvrent. Dans le cas des sorbants macroporeux, les pores jouent le rôle de canaux de transport. Les processus de condensation sont les plus significatifs dans les corps poreux transitoires. La condensation capillaire commence à certaines valeurs p et  lorsqu'une partie de l'énergie de surface a déjà été compensée. Une condition nécessaire est que la surface doit être autoportante. Le processus est décrit Équation de Thompson-Kelvin.
lorsqu'une partie de l'énergie de surface a déjà été compensée. Une condition nécessaire est que la surface doit être autoportante. Le processus est décrit Équation de Thompson-Kelvin.

 - pour le cas du mouillage, le centre de courbure est en phase gazeuse.
- pour le cas du mouillage, le centre de courbure est en phase gazeuse.
Dans le cas de la condensation capillaire, l'isotherme d'adsorption a une forme d'hystérésis. La branche inférieure correspond au processus d'adsorption et la branche supérieure correspond au processus de désorption.
Tous les types de pores peuvent être réduits à trois types :
|
conique |
Cylindrique avec une extrémité fermée |
Cylindrique à deux extrémités ouvertes |
|
Le remplissage du processus est effectué à partir du fond du pore. L'isotherme d'adsorption et l'isotherme de désorption coïncident dans ce cas, puisque le processus d'adsorption commence par une sphère et le processus de désorption commence également par la disparition de certaines sphères.
↓ |
Il n'y a pas d'hystérésis. Les courses avant et arrière sont décrites par l'équation :
|
Il n'y a pas de fond nulle part, le remplissage du pore ira le long des parois du cylindre.
cylindre: Isotherme et aura une forme d'hystérésis.
↓ |


 - sphère,
- sphère, ,
,



À  conditions de mouillage, la condensation se produit à des pressions plus basses, ce qui est énergétiquement favorable. A partir de la branche de désorption, des courbes de distribution de taille de pores sont obtenues.
conditions de mouillage, la condensation se produit à des pressions plus basses, ce qui est énergétiquement favorable. A partir de la branche de désorption, des courbes de distribution de taille de pores sont obtenues.
Le maximum de la courbe différentielle est décalé vers la gauche par rapport au point d'inflexion de l'intégrale. Le volume total des petits pores est petit, mais a de grandes surfaces. Plus la taille des pores augmente, plus leur volume augmente  , et la zone comme
, et la zone comme  , de ce fait, on observe un décalage du maximum de la courbe différentielle.
, de ce fait, on observe un décalage du maximum de la courbe différentielle.
Adsorption à l'interface solide-liquide.
Dans le cas de l'adsorption à l'interface solide-gaz, nous avons négligé un composant. Dans le cas de l'adsorption à l'interface solide-liquide, l'adsorbat déplace les molécules de solvant de la surface de l'adsorbant.
 ,
,
La bonne équation est :

 ,
,
N 1, N 2 - fractions molaires du solvant et du composant, N 1 + N 2 \u003d 1, puis
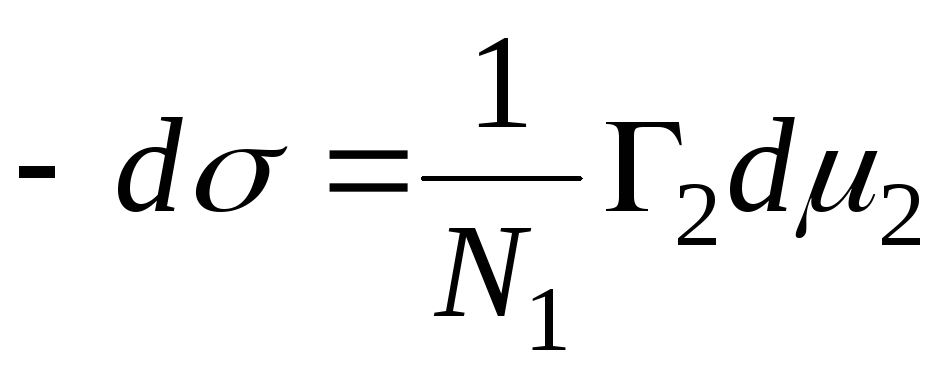 ,
=>
,
=>
 , puis - l'équation d'adsorption pour la limite de phase solide - liquide.
, puis - l'équation d'adsorption pour la limite de phase solide - liquide.
Adsorption (G) > 0 à  <
0
<
0
Si les valeurs  car le composant et le solvant sont très différents, dans ce cas la dépendance g de N a un extremum à la valeur N
~ 0,5.
car le composant et le solvant sont très différents, dans ce cas la dépendance g de N a un extremum à la valeur N
~ 0,5.
E  si
si  ont des valeurs similaires, dans ce cas le signe de l'adsorption peut changer. Dépendance g de N croise l'axe des x
ont des valeurs similaires, dans ce cas le signe de l'adsorption peut changer. Dépendance g de N croise l'axe des x
Intersection de fonctions g(N) avec l'axe des abscisses s'appelle azéotrope d'adsorption. Cela signifie que les deux composants ne peuvent pas être séparés sur cet adsorbant.
Équation isotherme d'adsorption avec constante d'échange.
Lors de l'adsorption à l'interface solide-liquide, les composants sont constamment redistribués entre la surface de l'adsorbant et le volume de la solution.
 - composants (- - se réfèrent à la surface)
- composants (- - se réfèrent à la surface)

 ,
,
 ,
, .
.
 ,
,


Adsorption à l'interface liquide-gaz
R  Considérons l'évolution du profil de concentration lors de la traversée de l'interface liquide-gaz. Soit le composant 2 volatil.
Considérons l'évolution du profil de concentration lors de la traversée de l'interface liquide-gaz. Soit le composant 2 volatil.
Cs est la concentration dans la couche de surface.
Basé sur la définition de l'excès d'adsorption

Si le composant n'est pas volatil, alors la valeur d'adsorption s'écrira comme suit :
P  ri
ri 

Dans l'équation  la nature de la matière est décrite par la dérivée
la nature de la matière est décrite par la dérivée  .
.
L'isotherme de tension superficielle peut être de la forme 1 ou 2 :
1 - tensioactifs
2 - tensioactifs
L'activité de surface g est la capacité des substances à réduire la tension de surface dans le système.

 - épaisseur de la couche superficielle
- épaisseur de la couche superficielle
C s est la concentration du composant dans la couche de surface
DE– concentration volumique
Pour une série homologue il existe une règle :
 - Règle Traubeau Duclos
- Règle Traubeau Duclos
Pour la série homologue, l'isotherme d'adsorption ressemble à ceci :
On note D au lieu de A, car l'adsorption est excessive dans la couche superficielle.
Isotherme de tension superficielle :

 est la tension superficielle du solvant pur.
est la tension superficielle du solvant pur.
 - équation fondamentale d'adsorption ;
- équation fondamentale d'adsorption ;
 - Équation de Langmuir.
- Équation de Langmuir.
Résolvons-les ensemble :

- L'équation de Chichkovski.
B est une constante pour la série homologue.
UN- lors du passage d'un homologue à un autre, il augmente de 3 à 3,5 fois 
![]()
1 - zone de faibles concentrations
![]()
2 - concentration moyenne
3 - couche monomoléculaire
Les tensioactifs sont des molécules amphiphiles, c'est-à-dire comprennent un groupe polaire et un radical hydrocarboné non polaire.
o est la partie polaire de la molécule.
| est la partie non polaire de la molécule.
Dans un solvant polaire, les molécules de tensioactif sont orientées de telle manière que la partie polaire de la molécule fait face au solvant, tandis que la partie non polaire est poussée dans la phase gazeuse.
Dans l'équation de Shishkovsky  , elle est constante pour la série homologue.
, elle est constante pour la série homologue.
L'action tensioactive commence à apparaître avec n>5. À des concentrations supérieures à la concentration de la couche monomoléculaire, la micellisation se produit dans les solutions de tensioactif.
Micellaire- on appelle l'agrégat de molécules tensioactives amphiphiles, dont les radicaux hydrocarbonés forment le noyau, et les groupements polaires sont transformés en phase aqueuse.
Masse micellaire - masse micellaire.
H  le nombre de molécules est le nombre d'agrégation.
le nombre de molécules est le nombre d'agrégation.
Micelles sphériques
Dans le cas de la micellisation, un équilibre s'établit dans la solution
CMC est la concentration micellaire critique.
Puisque nous considérons la micelle comme une phase distincte :
Pour la série homologique, il existe une équation empirique :
un est l'énergie de dissolution du groupe fonctionnel.
b est l'incrément de potentiel d'adsorption, le travail d'adsorption par unité de méthylène.
est l'incrément de potentiel d'adsorption, le travail d'adsorption par unité de méthylène.
La présence d'un noyau hydrocarboné dans les micelles permet aux composés insolubles dans l'eau de se dissoudre dans les solutions aqueuses de tensioactifs, ce phénomène est appelé solubilisation (ce qui se dissout est un solubilisant, le tensioactif est un solubilisant).
La boue peut être complètement non polaire, peut contenir à la fois des parties polaires et non polaires et sera orientée comme une molécule de surfactant.
Dans tous les cas, lors de la solubilisation, une augmentation de la masse micellaire et du nombre d'agrégation se produit non seulement en raison de l'inclusion du solubilisé, mais également en raison d'une augmentation du nombre de molécules de tensioactif nécessaires pour maintenir l'état d'équilibre.
La solubilisation est d'autant plus efficace que le poids moléculaire du solubilisé est faible.

 ~ 72 mN/m.
~ 72 mN/m.
 ~ 33 mN/m.
~ 33 mN/m.
L'efficacité des tensioactifs dépend de l'amplitude de la CMC.
Pression de la couche de surface 2D

→ -forces de tension superficielle.
- pression bidimensionnelle.
La couche superficielle est une force égale à la différence entre les tensions superficielles d'une solution de tensioactif et d'un solvant pur, dirigée vers une surface propre.
Un équilibre s'établit entre la solution et la couche superficielle



À  il y a un domaine où
il y a un domaine où  linéairement dépendant de la concentration.
linéairement dépendant de la concentration.
G [mol / m 2].
 surface occupée par une mole d'une substance
surface occupée par une mole d'une substance
Alors l'isotherme de pression bidimensionnelle aura la forme 
 est l'isotherme de pression bidimensionnelle.
est l'isotherme de pression bidimensionnelle.
Dépendance  de S M :
de S M :

À  - la pression bidimensionnelle augmente fortement. À
- la pression bidimensionnelle augmente fortement. À  bidimensionnel est déformé, ce qui provoque une forte croissance
bidimensionnel est déformé, ce qui provoque une forte croissance  .
.
Le film des deux côtés limité par les mêmes phases est appelé double face. Dans de tels films, un mouvement constant de la liqueur mère est observé.
Les films de moins de 5 nm d'épaisseur sont appelés films noirs.

Les couches d'adsorption doivent avoir deux caractéristiques : viscosité et mobilité aisée, fluidité et élasticité.
L'effet Marangoni s'auto-guérit.
Triangle de Gibbs,  - surpression.
- surpression.
Le film est étiré et du fait qu'une partie du liquide a disparu, les tensioactifs se précipitent dans l'espace libre. Triangle de Gibbs.
Effet de la force d'adsorption des corps.
Il y a toujours une couche d'adsorption à la surface du film, pour laquelle , alors
Équation de Langmuir :


 en pression bidimensionnelle
en pression bidimensionnelle
 - analogue de l'équation de Shishkovsky
- analogue de l'équation de Shishkovsky
phénomènes électrocinétiques. Double couche électrique (DES).
Modèle Helemholtz. Théorie de Gouy-Chapman.
Vol de 1808

tu – en forme de tube, plongeant dedans 2 électrodes. La loi des vases communicants est violée et il y a un changement du niveau de liquide dans le tube - phénomènes électrocinétiques.
Phénomènes cinétiques :
électrophorèse
Électrosmose
Potentiel de débit (débit)
Potentiel de sédimentation
1 et 2 surviennent lorsqu'une différence de potentiel est appliquée, 3 et 4 le poinçonnage et la sédimentation des particules colloïdales provoquent l'apparition d'une différence de potentiel.
Électrosmose est le déplacement d'un milieu de dispersion par rapport à une phase dispersée stationnaire sous l'action d'un courant électrique.
électrophorèse est le mouvement des particules de la phase dispersée par rapport à un milieu de dispersion fixe sous l'action d'un courant électrique.
P  La raison de l'apparition des phénomènes électrocinétiques est la séparation spatiale des charges et l'apparition d'une double couche électrique.
La raison de l'apparition des phénomènes électrocinétiques est la séparation spatiale des charges et l'apparition d'une double couche électrique.
La double couche électrique est un condensateur plat, une plaque est formée d'ions déterminant le potentiel, l'autre de contre-inoes. Les ions sont également contaminés car les co-ions déterminant le potentiel sont poussés dans la masse de la solution. Distance entre les plaques  . Le potentiel chute linéairement, la différence de potentiel
. Le potentiel chute linéairement, la différence de potentiel  .
.
Une différence de potentiel externe provoque l'apparition d'un module de cisaillement  est une paire de forces par unité de surface agissant le long de la surface d'un corps solide.
est une paire de forces par unité de surface agissant le long de la surface d'un corps solide.

A l'équilibre, le module de cisaillement est égal au module de frottement visqueux (  ).
).

Dans nos conditions  ,
,

 - Équation de Helemholtz-Smalukovsky
- Équation de Helemholtz-Smalukovsky
 - déplacement à vitesse linéaire i phases.
- déplacement à vitesse linéaire i phases.
E est l'intensité du champ électrique.
 - différence de potentiel entre les plaques
- différence de potentiel entre les plaques
 - mobilité électrophorétique [m 2 /(V * s)].
- mobilité électrophorétique [m 2 /(V * s)].
Le modèle d'Helemholtz ne tient pas compte du mouvement thermique des molécules. En réalité, la répartition des ions dans la double couche est plus complexe.
Gouy et Chapman ont identifié les causes suivantes du DES :
Passage d'un ion d'une phase à une autre lorsque l'équilibre est établi.
Ionisation de la matière en phase solide.
Complétion de la surface par des ions présents dans le milieu de dispersion.
Polarisation à partir d'une source de courant externe.
La double couche électrique a une structure floue ou diffuse. Les ions ont tendance à être uniformément répartis dans la couche diffuse.
La couche diffuse est constituée de contre-ions, la longueur de la couche est déterminée par leur énergie cinétique. A une température tendant vers le zéro absolu, les contre-inoes sont aussi proches que possible des ions déterminants du potentiel.
Cette théorie est basée sur deux équations :
Équation de Boltzmann

 - travailler contre les forces d'interaction électrostatique.
- travailler contre les forces d'interaction électrostatique.
 est la densité de charge apparente.
est la densité de charge apparente.

Équation de Poisson 
Étant donné que l'épaisseur DEL est beaucoup plus petite que la taille des particules, et pour un DEL plat, la dérivée par rapport aux coordonnées  et
et  est aboli.
est aboli. 
Pour e y avec y<<1 функцию можно разложить в ряд Маклорена:

On se limite à deux membres de la série, alors :


 - L'épaisseur DEL est la distance à laquelle le potentiel DEL diminue en e une fois que.
- L'épaisseur DEL est la distance à laquelle le potentiel DEL diminue en e une fois que.
Plus la température est basse, moins  . À Т→0 – DES plats. Plus la concentration est élevée, plus je, moins
. À Т→0 – DES plats. Plus la concentration est élevée, plus je, moins  .
.





 "-" signifie que le potentiel diminue avec la distance. =>
"-" signifie que le potentiel diminue avec la distance. =>
=>

 ,
,
 - le potentiel décroît exponentiellement.
- le potentiel décroît exponentiellement.
Potentiel de densité de charge de surface : 
La charge de surface est une charge d'espace de signe opposé, intégrée sur la distance.




=>


Où le potentiel diminue de 2,7 fois - 
Capacité double couche 
L'inconvénient de la théorie est que la présence de la couche d'Helemholtz n'est pas prise en compte, c'est-à-dire ne tient pas compte  , d'où les erreurs de détermination des principaux paramètres. Elle n'explique pas non plus l'effet des ions de nature différente sur l'épaisseur de la double couche électrique.
, d'où les erreurs de détermination des principaux paramètres. Elle n'explique pas non plus l'effet des ions de nature différente sur l'épaisseur de la double couche électrique.
La théorie de Stern. La structure d'une micelle colloïdale.
La double couche électrique est constituée de deux parties : dense et diffuse. Une couche dense se forme à la suite de l'interaction d'ions formant un potentiel avec des ions spécifiquement adsorbés. Ces ions, en règle générale, sont partiellement ou complètement déshydratés et peuvent avoir une charge identique ou opposée aux ions déterminant le potentiel. Cela dépend du rapport de l'énergie d'interaction électrostatique  et potentiel d'adsorption spécifique
et potentiel d'adsorption spécifique  . Les ions de la couche dense sont fixés. L'autre partie des ions est située dans la couche diffuse ; ces ions sont libres et peuvent pénétrer profondément dans la solution, c'est-à-dire d'une zone de concentration plus élevée à une zone de concentration plus faible. La densité de charge totale se compose de deux parties.
. Les ions de la couche dense sont fixés. L'autre partie des ions est située dans la couche diffuse ; ces ions sont libres et peuvent pénétrer profondément dans la solution, c'est-à-dire d'une zone de concentration plus élevée à une zone de concentration plus faible. La densité de charge totale se compose de deux parties.

 - Charge de couche Helmholtz
- Charge de couche Helmholtz
 -Charge de la couche diffuse
-Charge de la couche diffuse
La surface possède un certain nombre de centres d'adsorption qui interagissent chacun avec un contre-ion. La constante d'une telle réaction quasi-chimique est :

 , où
, où  - fraction molaire des contre-ions en solution
- fraction molaire des contre-ions en solution
Répartition de Helmholtz
Le potentiel décroît linéairement
Distribution potentielle de Gouy. Il n'y a pas de couche dense, le potentiel décroît exponentiellement à partir de la valeur 
Répartition arrière.
Au départ, la décroissance du potentiel est linéaire, puis exponentielle.
Lorsqu'un champ électrique est appliqué dans le cas de l'électrophorèse, ce n'est pas la particule de la phase solide qui se déplace directement, mais la particule de la phase solide avec une couche d'ions environnante. Le DES reprend la forme de la particule de la phase dispersée. Lorsqu'un potentiel est appliqué, une partie de la couche diffuse est arrachée. La ligne de rupture s'appelle limite coulissante.
Le potentiel qui apparaît à la limite de glissement à la suite du détachement d'une partie de la couche diffuse est appelé potentiel électrocinétique(Potentiel Zeta  ).
).
Une particule d'une phase dispersée, entourée d'une couche de contre-ions et d'une double couche électrique, est appelée micelle.
Règles d'écriture des micelles colloïdales :


1-1 électrolyte de charge
T est une particule de la phase dispersée.
AA est la frontière entre les parties dense et diffuse.
BB est la limite de glissement.
La limite de glissement peut ou non coïncider avec la ligne AA.
La valeur de pH à laquelle le potentiel zêta est nul est appelée point isoelectrique.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. En excès de CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n-x)Cl - ) 2 X + X Cl - - enregistrer les micelles.
CaSO 4 m - agrégat.
CaSO 4 m∙nCa 2+ est le noyau.
CaSO 4 m∙nCa 2+ 2( n-x)Cl - particule.
2. En excès de Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micelle
CaSO 4 m - agrégat.
CaSO 4 m∙nSO 4 2 + est le noyau.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - particule
Équation de Helemholtz-Smoluchowski

 - vitesse linéaire de déplacement des joints (en électroosmose).
- vitesse linéaire de déplacement des joints (en électroosmose).
 - différence de potentiel sur les armatures du condensateur (en électroosmose).
- différence de potentiel sur les armatures du condensateur (en électroosmose).


 - débit volumétrique de la solution, S est la section transversale de la cellule.
- débit volumétrique de la solution, S est la section transversale de la cellule.

E est l'intensité du champ électrique.


 (pour l'électroosmose).
(pour l'électroosmose).
Pour le potentiel d'écoulement :

 - potentiel
- potentiel
 - pression membranaire
- pression membranaire
En règle générale, la valeur des mobilités électrophorétiques et des mobilités électroosmotiques est inférieure à celles calculées. Cela est dû à:
Effet de relaxation (lors du mouvement d'une particule de la phase dispersée, la symétrie de l'atmosphère ionique est violée).
Freinage électrophorétique (l'apparition d'un frottement supplémentaire à la suite du mouvement des contre-ions).
Distorsion des lignes de courant dans le cas de particules électriquement conductrices.
Relation entre la tension superficielle et le potentiel. Équation de Lippmann.
La formation de DEL se produit spontanément en raison de la volonté du système de réduire son énergie de surface. Dans le cadre de la constance J et p l'équation généralisée des première et deuxième lois de la thermodynamique ressemble à :
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1ère équation de Lippmann.
- 1ère équation de Lippmann.
 est la densité de charge de surface.
est la densité de charge de surface.
 - capacité différentielle.
- capacité différentielle.
 - 2ème équation de Lippmann.
- 2ème équation de Lippmann.
DE- capacité.
On résout la 1ère équation de Lippmann et l'équation fondamentale d'adsorption :
 ,
,


 , alors
, alors
 - Équation de Nernst
- Équation de Nernst
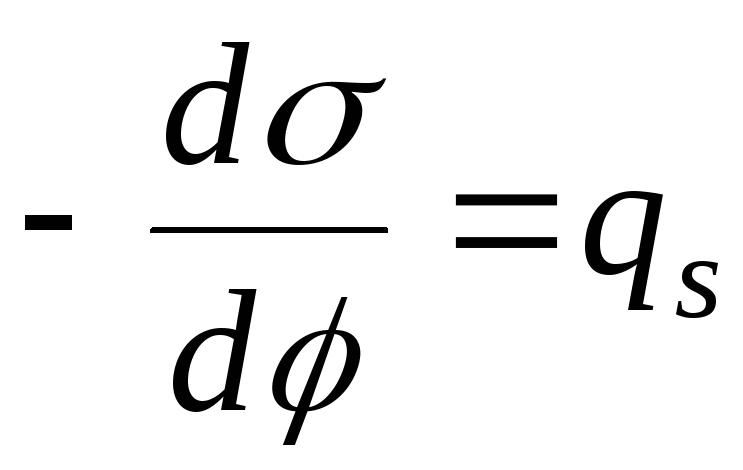 ,
,
 ,
,




 - l'équation de la courbe électrocapillaire (ECC).
- l'équation de la courbe électrocapillaire (ECC).
À  :
: , mais
, mais 
Les tensioactifs cationiques (CSAS) réduisent la branche cathodique de l'ECC.
Les tensioactifs anioniques (ASS) réduisent la branche anodique de l'ECC.
Les tensioactifs non ioniques (NSA) réduisent la partie médiane de l'ECC.
Stabilité des systèmes dispersés. Pression de calage.
Les systèmes dispersés peuvent être divisés :
Les systèmes thermodynamiquement instables peuvent être cinétiquement stables en raison de la transition vers un état métastable.
Il existe deux types de stabilité :
Stabilité de la sédimentation (par rapport à la gravité).
Stabilité agrégative. (par rapport au collage)
Coagulation est le processus par lequel les particules se collent les unes aux autres, entraînant la perte de stabilité agrégative. La coagulation peut être causée par des changements de température, de pH, d'agitation, d'ultrasons.
Distinguer la coagulation :
Réversible.
Irréversible.
La coagulation se poursuit avec l'introduction d'électrolytes.
Règles de coagulation :

Film- C'est la partie du système située entre deux interfaces.
pression disjointe se produit avec une forte diminution de l'épaisseur du film en raison de l'interaction des couches de surface qui s'approchent.

«-» - lorsque l'épaisseur du film diminue, la pression de disjonction augmente.
P 0 est la pression dans la phase brute, qui est un prolongement de l'intercalaire.
P 1 est la pression dans le film.
Théorie de la stabilité. DLFO (Deryagin, Landau, Fairway, Overbeck).
Selon la théorie DLVO, deux composantes se distinguent dans la pression disjointe :
électrostatique P E (positif, il est dû aux forces de répulsion électrostatique). Correspond à une diminution de l'énergie de Gibbs avec l'augmentation de l'épaisseur du film.
Moléculaire PM (négatif, dû à l'action des forces attractives). Elle est provoquée par la compression du film due aux forces chimiques de surface, le rayon d'action des forces est du dixième de nm avec une énergie de l'ordre de 400 kJ/mol.
Énergie d'interaction totale:

 - le système est globalement stable
- le système est globalement stable
 - système instable
- système instable
P  composante positive.
composante positive.
L'augmentation est due à l'augmentation de l'énergie potentielle lors de la compression des couches minces. Pour les couches épaisses, l'excès d'énergie ionique est compensé et est égal à l'énergie d'interaction dans la masse du milieu de dispersion.
Si un  (
( - l'épaisseur du film,
- l'épaisseur du film,  - rayon de l'ion) l'amincissement du film entraîne la disparition et la réduction dans celui-ci de molécules et d'ions avec une énergie de surface minimale. Le nombre de particules voisines diminue, ce qui entraîne une augmentation de l'énergie potentielle des particules restant dans le film.
- rayon de l'ion) l'amincissement du film entraîne la disparition et la réduction dans celui-ci de molécules et d'ions avec une énergie de surface minimale. Le nombre de particules voisines diminue, ce qui entraîne une augmentation de l'énergie potentielle des particules restant dans le film.


La théorie DLVO considère l'interaction des particules comme l'interaction des plaques.

Les particules n'interagissent pas



 - équation de Laplace,
- équation de Laplace,  ,
,


Pour les surfaces faiblement chargées
Pour les surfaces fortement chargées :

La composante moléculaire est l'interaction de deux atomes :
 ~
~
Interaction d'un atome avec une surface :

Prenons deux enregistrements :
ré  Pour obtenir la composante moléculaire, il faut sommer toutes les énergies d'interaction des atomes des plaques droite et gauche.
Pour obtenir la composante moléculaire, il faut sommer toutes les énergies d'interaction des atomes des plaques droite et gauche.
où  - Constante de Hamaker (prend en compte la nature des corps en interaction).
- Constante de Hamaker (prend en compte la nature des corps en interaction).
Ce. l'énergie d'interaction des particules dans un système peut être exprimée à l'aide de courbes de potentiel.
I est le minimum de potentiel primaire. C'est une zone de coagulation irréversible, les forces d'attraction prédominent.
II - zone de stabilité agrégative, les forces répulsives prévalent.
III - minimum de potentiel secondaire (ou zone de floculation). Entre les particules de la phase dispersée se trouve une couche d'électrolyte, et les particules peuvent être séparées et transférées vers la zone de stabilité d'agrégation.

Courbe 1 – le système est globalement stable.
La courbe 2 est stable en zone I, non stable en zone II.
Courbe 3 - la coagulation s'est produite dans le système.
Courbe 4 - au point 4, l'énergie totale d'interaction U=0,  , ce point extremum correspond au début d'une coagulation rapide.
, ce point extremum correspond au début d'une coagulation rapide.
Il y a deux cas :
1. Les surfaces sont faiblement chargées :
U \u003d U E + U M \u003d 0
 (1)
(1)
2)

 (2)
(2)



 - c'est l'épaisseur de la couche correspondant au début du processus de coagulation.
- c'est l'épaisseur de la couche correspondant au début du processus de coagulation.






 - pour les surfaces faiblement chargées
- pour les surfaces faiblement chargées
 alors
alors 
2. Pour les surfaces fortement chargées :
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Faisons le carré (3)

Coagulation:
Dans l'adsorption spécifique, les ions peuvent être adsorbés en une quantité suréquivalente de telle sorte que la surface peut changer sa charge. La surface se recharge.
Dans le cas d'une adsorption spécifique, non seulement des ions de signes opposés, mais également un ion peuvent être adsorbés.
Si des ions de même signe que la surface sont adsorbés, alors dans la couche de surface, il n'y aura pas de baisse du potentiel, mais sa croissance.

Coagulation de neutralisation (se produit avec la participation de particules faiblement chargées et dépend non seulement de la charge de l'électrolyte coagulant, mais également du potentiel à la frontière des couches denses et diffuses).
Théorie de la coagulation rapide de Smoluchowski.

Dépendance du taux de coagulation sur la concentration d'électrolyte.
I – le taux de coagulation est faible,
II - la vitesse de coagulation est pratiquement proportionnelle à la concentration en électrolyte.
III - la zone de coagulation rapide, le taux est pratiquement indépendant de la concentration.
Points clés:
Le sol initial est monodisperse, les particules similaires ont une forme sphérique.
Toutes les collisions de particules sont efficaces.
Lorsque deux particules primaires entrent en collision, une particule secondaire se forme. Secondaire + primaire = tertiaire. Primaire, secondaire, tertiaire - multiplicité.
En termes de cinétique chimique, le processus de coagulation peut être décrit par l'équation :

La solution sera l'équation : 
 - temps de demi-coagulation. C'est le temps pendant lequel le nombre de particules de sol diminue de 2 fois.
- temps de demi-coagulation. C'est le temps pendant lequel le nombre de particules de sol diminue de 2 fois.

 ,
,
 ,
,
 ,
,


Lorsque la multiplicité augmente, le maximum des courbes de coagulation se déplace vers des valeurs plus grandes  .
.
Défauts:
Hypothèse de monodispersité.
L'hypothèse sur l'efficacité de toutes les collisions.
Nous recommandons également
 Passage du jeu Darksiders II Key to Redemption
Passage du jeu Darksiders II Key to Redemption
 Procédure pas à pas complète des quêtes principales et secondaires du deuxième chapitre
Procédure pas à pas complète des quêtes principales et secondaires du deuxième chapitre
 Bob l'éponge SquarePants (Soluce pas à pas)
Bob l'éponge SquarePants (Soluce pas à pas)
 jeux de bob l'éponge
jeux de bob l'éponge
 Escape Impossible: Revenge Procédure pas à pas Procédure pas à pas Pouvez-vous vous échapper de la vidéo
Escape Impossible: Revenge Procédure pas à pas Procédure pas à pas Pouvez-vous vous échapper de la vidéo
 Et le dragon est venu. Difficulté de choix. Points clés du jeu The Witcher 2 tuer le dragon ou échapper aux conséquences
Et le dragon est venu. Difficulté de choix. Points clés du jeu The Witcher 2 tuer le dragon ou échapper aux conséquences