Mišična distrofija: vzroki, simptomi in zdravljenje. progresivne mišične distrofije
Odlikujeta jih selektivna porazdelitev šibkosti in specifična narava genetskih nepravilnosti.
Beckerjeva distrofija, čeprav je tesno povezana, se pojavi pozneje in povzroča blažje simptome. Druge oblike vključujejo Emery-Dreyfusovo distrofijo, miotonično distrofijo, distrofijo pasu okončin, faciokapulohumeralno distrofijo in prirojene distrofije.
Duchennova mišična distrofija in Beckerjeva mišična distrofija
Duchennova mišična distrofija in Beckerjeva mišična distrofija sta X-povezani recesivni motnji, za katere je značilna progresivna oslabelost proksimalnih mišic, ki jo povzroča degeneracija mišičnih vlaken. Beckerjeva distrofija se pojavi pozno in povzroča blažje simptome. Zdravljenje se osredotoča na ohranjanje funkcije s fizikalno terapijo, naramnicami in ortozami; Prednizolon je predpisan nekaterim bolnikom s hudo funkcionalno okvaro.
Pri Duchennovi distrofiji ta mutacija povzroči hudo odsotnost (<5%) дистрофина, белка мембраны мышечных клеток. При дистрофии Беккера мутация приводит к образованию ненормального дистрофина или малому его количеству. Дистрофия Дюшенна поражает 1/3000 родившихся мужчин. Дистрофию Беккера выявляют у 1/30 000 родившихся мужчин. У женщин-носителей может быть выражено бессимптомное повышение уровня креатинкиназы и, возможно, гипертрофия задней части голени.
Simptomi in znaki
Duchennova distrofija. Ta motnja se običajno kaže v starosti 2-3 let. Slabost prizadene proksimalne mišice, običajno najprej v spodnjih okončinah. Otroci pogosto hodijo po prstih, imajo galajočo hojo in lordozo. Napredovanje šibkosti je enakomerno, razvijajo se fleksijske kontrakture okončin in skolioza. Razvija se izrazita psevdohipertrofija. Večina otrok je privezanih na invalidski voziček do 12. leta. Prizadetost srca je običajno asimptomatska, čeprav ima 90 % bolnikov nenormalnosti EKG. Ena tretjina jih ima blage, neprogresivne motnje v duševnem razvoju, ki vplivajo na verbalne sposobnosti bolj kot na uspešnost.
Beckerjeva distrofija. Ta motnja se običajno kaže simptomatsko veliko kasneje in je blažja. Sposobnost gibanja običajno traja vsaj do 15. leta starosti, veliko otrok pa ostane mobilnih tudi v odrasli dobi. Večina prizadetih živi v 30-ih in 40-ih letih.
Diagnostika
- Imunsko barvanje z distrofinom. i Analiza DNK za mutacije.
Sum na diagnozo temelji na značilnih kliničnih značilnostih, starosti začetka in družinski anamnezi, ki kaže na X-povezano recesivno dedovanje. Miopatske spremembe so vidne na elektromiografiji (potenciali motoričnih enot hitro naraščajo, so kratkotrajni in nizke amplitude) in pri biopsiji mišic (nekroza in izrazita sprememba velikosti mišičnih vlaken, ki niso ločena od motoričnih enot). Ravni kreatin kinaze so presegle 100-krat normalno.
Diagnozo potrdimo z analizo distrofina z imunskim barvanjem biopsijskih vzorcev. Distrofija pri bolnikih z Duchennovo distrofijo ni odkrita. Tudi analiza mutacij DNK v levkocitih periferne krvi lahko potrdi diagnozo, ko se odkrijejo nepravilnosti v genu za distrofin (približno 65 % bolnikov ima delecije ali podvojene, približno 25 % pa točkovne mutacije).
Zdravljenje
- podporne ukrepe.
- Včasih prednizon.
- Včasih korektivna operacija.
Specifičnega zdravljenja ni. Zmerna vadba je priporočljiva čim dlje. Opornica za gleženj bo pomagala preprečiti upogibanje med spanjem. Ortopedski pripomočki na nogah lahko začasno pomagajo ohranjati zmožnost vstajanja in gibanja. Izogibati se je treba debelosti; potrebe po kalorijah bodo verjetno nižje kot običajno. Indicirano je genetsko svetovanje.
Dnevna uporaba prednizona ne povzroči pomembnega dolgoročnega kliničnega izboljšanja, lahko pa upočasni potek bolezni. Glede dolgoročne učinkovitosti ni soglasja. Genska terapija še ni razvita. Včasih je potrebna korekcijska operacija. Odpoved dihanja je včasih mogoče zdraviti z neinvazivno respiratorno podporo (prek nosne maske). Elektivna traheotomija postaja vse bolj sprejemljiva, kar otrokom z Duchennovo distrofijo omogoča preživetje do 20. leta.
Druge oblike mišične distrofije
Emery-Dreyfusova distrofija. Ta bolezen se lahko deduje avtosomno dominantno, avtosomno recesivno (najredkejša) ali X-vezan vzorcu. Celotna frekvenca ni znana. Samice so lahko nosilke, vendar so le samci klinično prizadeti z dedovanjem, vezanim na X. Geni, povezani z Emery-Dreyfusovo distrofijo, kodirajo proteina jedrske membrane lamin A/C (avtosomno) in emerin (povezan z X).
Simptomi mišične oslabelosti in izgubljanja se lahko pojavijo kadar koli pred 20. letom in običajno prizadenejo bicepse, tricepse in redkeje distalne mišice nog. Srce je pogosto povezano z atrijsko fibrilacijo, motnjami prevodnosti (atrioventrikularni blok), kardiomiopatijo in veliko verjetnostjo nenadne smrti.
Na diagnozo kažejo klinični izvidi, starost začetka in družinska anamneza. Kot tudi rahlo povišane ravni kreatin kinaze v serumu in miopatski znaki na elektromiografiji in mišični biopsiji. Diagnozo potrdi DNK testiranje.
Zdravljenje vključuje terapijo, ki je namenjena preprečevanju kontraktur. Srčni spodbujevalniki so včasih ključnega pomena pri bolnikih z nenormalno prevodnostjo.
Miotonična distrofija. Miotonična distrofija je najpogostejša oblika mišične distrofije pri beli populaciji. Pojavlja se s pogostostjo približno 30/100.000 živorojenih moških in žensk. Dedovanje je avtosomno dominantno z različno penetracijo. Dva genetska lokusa - DM 1 in DM 2 - povzročata nenormalnosti. Simptomi in znaki se začnejo v adolescenci ali adolescenci in vključujejo miotonijo (zakasnjena sprostitev po krčenju mišic), šibkost in izčrpanost distalnih mišic okončin (zlasti rok) in obraznih mišic (zlasti pogosta je ptoza) ter kardiomiopatijo. Lahko se razvijejo tudi duševna zaostalost, katarakta in endokrine motnje.
Diagnozo kažejo značilni klinični izvidi, starost začetka in družinska anamneza; Diagnozo potrdi DNK testiranje. Zdravljenje vključuje uporabo ortoze za povešeno stopalo in zdravljenje z zdravili za miotonijo (npr. meksiletin 75–150 mg peroralno 2–3 krat na dan).
Distrofija pasov okončin. Trenutno je znanih 21 podtipov distrofije pasu okončin: 15 avtosomno recesivnih in 6 avtosomno dominantnih. Celotna frekvenca ni znana. Ugotovljenih je bilo več kromosomskih lokusov za avtosomno dominantne (5q [genski produkt neznan)] in recesivne (2q, 4q [, 13q [γ-sarkoglikan], 15Q [kalpain, Ca-aktivirane proteaze] in 17q [α-sarkoglikan ali adhalin] ) obrazci. Lahko so prizadeti strukturni (npr. z distrofinom povezani glikoproteini) ali nestrukturni (npr. proteaze) proteini.
Simptomi vključujejo šibkost v pasu in proksimalnih okončinah. Začetek bolezni sega od zgodnjega otroštva do odraslosti; Začetek avtosomno recesivnih tipov je običajno v otroštvu in te vrste so večinoma povezane s prizadetostjo medeničnega obroča.
Diagnozo kažejo značilni klinični izvidi, starost začetka in družinska anamneza; Diagnoza zahteva tudi določitev histološke slike mišic, imunocitokemijo, Western blotting in genetsko testiranje na prisotnost specifičnih beljakovin.
Zdravljenje je namenjeno preprečevanju kontraktur.
Facioskapulohumeralna distrofija. Za nastanek bolezni v adolescenci ali adolescenci je značilno počasno napredovanje: otrok ima težave s žvižganjem, zapiranjem oči in dvigovanjem rok (zaradi oslabelosti mišic, ki stabilizirajo lopatice). Pričakovana življenjska doba je normalna. Infantilne variacije, za katere je značilna šibkost obraza, ramen in kolčnega pasu, hitro napredujejo.
Diagnozo kažejo značilni klinični izvidi, starost začetka in družinska anamneza; Diagnozo potrdi DNK analiza.
Zdravljenje je sestavljeno iz fizikalne terapije.
prirojena mišična distrofija. Ne gre za samostojno motnjo, temveč za prirojeno motnjo, ki je ena od redkih oblik mišične distrofije. Na diagnozo sumimo pri katerem koli mlahavnem novorojenčku, vendar jo je treba razlikovati od prirojene miopatije z mišično biopsijo.
Zdravljenje je fizikalna terapija, ki lahko pomaga ohraniti delovanje mišic.
Mišična distrofija je pravzaprav skupina dednih bolezni, za katere je značilna progresivna simetrična atrofija skeletnih mišic, ki se pojavlja brez bolečin in izgube občutljivosti v okončinah. Paradoksalno je, da se prizadete mišice lahko povečajo zaradi rasti vezivnega tkiva in maščobnih oblog, kar daje napačen vtis močnih mišic.
Do zdaj ni zdravila za mišično distrofijo. Obstajajo štiri glavne vrste te patologije. Najpogostejša je Duchennova mišična distrofija (50 % vseh primerov). Bolezen se običajno začne v zgodnjem otroštvu in vodi v smrt do 20. leta. Beckerjeva mišična distrofija se razvija počasneje, bolniki živijo več kot 40 let. Distrofija ramenskega obraza in okončin običajno ne vpliva na pričakovano življenjsko dobo.
Vzroki
razvoj mišične distrofije. zaradi različnih genov. Duchennovo mišično distrofijo in Beckerjevo mišično distrofijo povzročajo geni, ki jih najdemo na spolnem kromosomu in prizadeneta samo moške. Ramensko-skapularno-obrazne in udovno-ledvene distrofije niso povezane s spolnimi kromosomi; prizadenejo tako moške kot ženske.
Simptomi
Vse vrste mišične distrofije povzročajo progresivno mišično atrofijo.
Diagnostika
Zdravnik pregleda otroka, sprašuje o boleznih družinskih članov in predpiše določene študije. Če je kdo od sorodnikov zbolel za mišično distrofijo, zdravnik ugotovi, kako je potekala njegova distrofija. Z analizo pridobljenih podatkov je mogoče predvideti, kaj otrok pričakuje. Če v družini ni bilo bolnikov z mišično distrofijo, bo elektromiografija ocenila delovanje živcev v prizadetih mišicah in ugotovila prisotnost mišične distrofije; pregled koščka mišičnega tkiva () lahko pokaže celične spremembe in prisotnost telesne maščobe.
V zdravstvenih centrih, opremljenih z najsodobnejšo opremo za molekularno biologijo in imunološke raziskave, lahko natančno ugotovijo, ali bo otrok zbolel za mišično distrofijo. Ti centri lahko testirajo tudi starše in sorodnike na prisotnost genov, ki določajo razvoj Duchennove mišične distrofije in Beckerjeve mišične distrofije.
Vrste bolezni
Glede na resnost bolezni in čas njenega pojava so:
Duchennova distonija se kaže že v zgodnji starosti (od 3 do 5 let). Bolni otroci se preganjajo, se s težavo vzpenjajo po stopnicah, pogosto padejo in ne morejo teči. Ko dvignejo roke, njihove lopatice "zaostajajo" za trupom - ta simptom se imenuje "pterigoidne lopatice". Običajno je otrok z mišično distrofijo priklenjen na invalidski voziček do starosti 9-12 let. Progresivna šibkost srčne mišice vodi v smrt zaradi nenadnega srčnega popuščanja, odpovedi dihanja ali okužbe.
Čeprav ima Beckerjeva distrofija veliko skupnega z Duchennovo distrofijo, se razvija veliko počasneje. Simptomi se pojavijo pri starosti približno 5 let, po 15 letih pa prizadeti otroci običajno še vedno ohranijo sposobnost hoje, včasih pa tudi veliko kasneje.
Ramensko-skapularno-obrazna distrofija se razvija počasi, njen potek je relativno benigen. Najpogosteje se začne pred 10. letom starosti, lahko pa se pojavi v zgodnjem adolescenci. Otroci, ki imajo pozneje to patologijo, v otroštvu slabo sesajo; ko postanejo starejši, jim ne uspe stisniti ustnic kot piščalka, dvigniti roke nad glavo. Pri bolnih otrocih se obrazi razlikujejo po nedejavnosti pri smehu ali joku, včasih opazimo izraze obraza, ki se razlikujejo od običajnih.
Pacientova dejanja
Če vas skrbi, da bi vaš otrok morda razvil mišično distrofijo, boste morda želeli prinesti fotografije ali videoposnetke, ki ponazarjajo vaše posebne skrbi. S seboj pripeljite sorodnika ali prijatelja, ki bo prav tako prisluhnil informacijam zdravnika.
Zdravljenje
Do zdaj ni nobenega zdravila, ki bi lahko ustavilo napredovanje mišične atrofije pri mišični distrofiji. Vendar pa lahko ortopedski pripomočki, pa tudi vadbena terapija, fizikalna terapija in kirurški posegi za odpravo kontraktur lahko otroka ali najstnika za nekaj časa zadržijo v mobilnosti.
Družinski člani z anamnezo mišične distrofije morajo poiskati zdravniško genetsko svetovanje, da ugotovijo, ali obstaja tveganje za prenos bolezni na nerojenega otroka.
Zapleti
Nekatere vrste mišične distrofije skrajšajo življenjsko dobo osebe in pogosto prizadenejo mišice, povezane z dihanjem. Tudi z izboljšanim mehanskim dihanjem ljudje, ki imajo Duchennovo mišično distrofijo – najpogostejšo vrsto mišične distrofije – ponavadi umrejo zaradi odpovedi dihanja, preden dosežejo 40 let.
Številne vrste mišične distrofije lahko zmanjšajo tudi učinkovitost srčne mišice. Če so prizadete mišice, povezane s požiranjem, se lahko pojavijo težave s prehranjevanjem.
Ko mišična oslabelost napreduje, postane mobilnost težava. Mnogi ljudje, ki razvijejo mišično distrofijo, bodo morda morali uporabljati invalidski voziček. Vendar pa lahko dolgotrajna nepremičnost sklepov, povezana z uporabo invalidskega vozička, poslabša kontrakturo, pri kateri se okončine vrtijo in zaklenejo v notranji položaj.
Kontrakture lahko igrajo tudi vlogo pri razvoju skolioze, saj povzročijo ukrivljenost hrbtenice, kar dodatno zmanjša učinkovitost pljuč pri ljudeh z mišično distrofijo.
Preprečevanje
Ker lahko okužbe dihal postanejo težava v kasnejših fazah mišične distrofije, je pomembno, da se cepite proti pljučnici in redno cepite proti gripi.
Duchennova mišična distrofija je genetska bolezen, povezana s kršitvijo strukture mišičnih vlaken. Mišična vlakna pri tej bolezni sčasoma razpadejo in sposobnost gibanja se izgubi. Duchennova mišična distrofija je spolno povezana in prizadene moške. Pojavlja se že v otroštvu. Poleg mišičnih motenj bolezen vodi do deformacij skeleta, spremljajo jo lahko dihalno in srčno popuščanje, duševne in endokrine motnje. Za zdaj še ni radikalne metode zdravljenja za izkoreninjenje bolezni. Vsi obstoječi ukrepi so le simptomatski. Zelo redko bolnikom uspe preživeti mejnik 30 let. Ta članek se osredotoča na vzroke, simptome, diagnozo in zdravljenje Duchennove mišične distrofije.
Bolezen je leta 1861 (po drugih virih - 1868) prvič opisal francoski nevrolog in nosi njegovo ime. Ni tako redko: 1 primer na 3500 novorojenčkov. Od vseh mišičnih distrofij, ki jih pozna medicina, je ta najpogostejša.

Duchennova mišična distrofija temelji na genetski okvari na spolnem X kromosomu.
En del kromosoma X vsebuje gen, ki kodira proizvodnjo posebnega mišičnega proteina, imenovanega distrofin, v telesu. Protein distrofin je osnova mišičnih vlaken (miofibril) na mikroskopski ravni. Funkcija distrofina je vzdrževanje celičnega okostja, zagotavljanje sposobnosti miofibril, da izvajajo ponavljajoča se dejanja krčenja in sprostitve. Pri Duchennovi mišični distrofiji je ta beljakovina v celoti odsotna ali pa je sintetizirana pomanjkljivo. Raven normalnega distrofina ne presega 3%. To vodi do uničenja mišičnih vlaken. Mišice se ponovno rodijo in nadomestijo z maščobnim in vezivnim tkivom. Seveda se v tem primeru izgubi motorična komponenta človeške dejavnosti.
Bolezen se podeduje kot recesivni tip, povezan s kromosomom X. Kaj to pomeni? Ker so vsi človeški geni parni, torej se med seboj podvajajo, je za pojav patoloških sprememb v telesu z dedno boleznijo nujno, da pride do genetske napake v enem kromosomu ali podobnih delih obeh kromosomov. Če se bolezen pojavi le z mutacijami v obeh kromosomih, se ta vrsta dedovanja imenuje recesivna. Kadar se genetska anomalija odkrije samo v enem kromosomu, vendar se bolezen še vedno razvija, se ta vrsta dedovanja imenuje dominantna. Recesivni tip je možen le s hkratnim porazom enakih kromosomov. Če je drugi kromosom "zdrav", potem do bolezni ne bo prišlo. Zato je Duchennova mišična distrofija veliko moških, saj imajo en kromosom X v genetskem naboru, drugi (parni) Y. Če fant naleti na "zlomljen" kromosom X, bo zagotovo imel bolezen. , ker zdravega kromosoma pač nima. Da bi se pri deklici pojavila Duchennova mišična distrofija, se morata v njenem genotipu ujemati dva patološka kromosoma X, kar je praktično malo verjetno (v tem primeru mora biti deklicin oče bolan, materin genetski niz pa mora vsebovati okvarjen X kromosom) . Dekleta delujejo le kot prenašalci bolezni in jo prenašajo na svoje sinove. Seveda nekateri primeri bolezni niso posledica dedovanja, ampak se pojavljajo sporadično. To pomeni pojav mutacije v otrokovi genetski sestavi spontano. Novo nastala mutacija je lahko podedovana (pod pogojem, da je ohranjena sposobnost razmnoževanja).
Simptomi bolezni
Duchennova mišična distrofija se vedno pokaže pred 5. letom starosti. Najpogosteje se prvi simptomi pojavijo pred 3. letom starosti. Vse patološke manifestacije bolezni lahko razdelimo v več skupin (odvisno od narave sprememb):
- poškodbe skeletnih mišic;
- deformacije skeleta;
- poškodbe srčne mišice;
- duševne motnje;
- endokrine motnje.
Poškodbe skeletnih mišic

Poškodba mišičnega tkiva je glavna manifestacija bolezni. Povzroča splošno mišično oslabelost. Začetni simptomi se neopazno prikradejo.
Otroci se rodijo brez posebnih odstopanj. Vendar njihov motorični razvoj zaostaja v tempu v primerjavi z vrstniki. Takšni otroci so motorično manj aktivni in mobilni. Medtem ko je otrok zelo majhen, je to pogosto povezano s posebnostmi temperamenta in ne bodite pozorni na začetne spremembe.
Z začetkom hoje se pojavijo očitni znaki. Otroci pogosto padajo in se premikajo na prste (na prste). Opozoriti je treba, da se te kršitve ne razlagajo pri prvih otrokovih korakih, saj je dvonožno gibanje na začetku pri vseh otrocih povezano s padci in nerodnostjo. Ko se večina njihovih vrstnikov že giblje precej samozavestno, fantje z Duchennovo mišično distrofijo trmasto padajo.
Ko se otrok nauči govoriti, se začne pritoževati zaradi šibkosti in utrujenosti, nestrpnosti do fizičnega napora. Tek, plezanje, skakanje in druge najljubše dejavnosti otrok za otroka z Duchennovo mišično distrofijo niso privlačne.
Hoja takšnih otrok je podobna raci: zdi se, da se prevračajo z noge na nogo.
Posebna manifestacija bolezni je Gowersov simptom. Sestoji iz naslednjega: ko se otrok skuša dvigniti s kolen, počepov, s tal, z rokami pomaga šibkim mišicam nog. Da bi to naredil, se nasloni z rokami nase, "se povzpne po lestvi, sam."
Duchennova mišična distrofija je naraščajoča vrsta mišične oslabelosti. To pomeni, da se šibkost najprej pokaže v nogah, nato se razširi na medenico in trup, nato na ramena, vrat in na koncu na roke, dihalne mišice in glavo.
Kljub dejstvu, da se pri tej bolezni mišična vlakna uničijo in atrofirajo, so lahko navzven nekatere mišice videti povsem normalne ali celo napihnjene. Razvija se tako imenovana psevdohipertrofija mišic. Najpogosteje je ta proces opazen v teletih, glutealnih in deltoidnih mišicah, mišicah jezika. Mesto razpadlih mišičnih vlaken zaseda maščobno tkivo, zato se ustvari učinek dobrega razvoja mišic, ki se izkaže za popolnoma drugačen od testa.
Atrofični proces v mišicah je vedno simetričen. Smer procesa navzgor vodi do videza "ose" pasu, "krilastih" lopatic (lopatice zaostajajo za telesom kot krila), simptoma "ohlapnega ramenskega obroča" (ko se zdi, da se glava padejo v ramena, ko poskušate otroka dvigniti pod pazduho). Obraz je hipomimičen, ustnice se lahko zadebelijo (nadomestitev mišic z maščobnim in vezivnim tkivom). Psevdohipertrofija jezika postane vzrok za motnje govora.
Uničenje mišic spremlja razvoj mišičnih kontraktur in skrajšanje kit (jasno razvidno na primeru Ahilove tetive).
Tetivni refleksi (koleni, ahil, biceps, triceps itd.) se postopoma zmanjšujejo. Mišice so na dotik čvrste, vendar neboleče. Mišični tonus se običajno zmanjša.
Postopno napredovanje mišične oslabelosti vodi v dejstvo, da do starosti 10-12 let mnogi otroci izgubijo sposobnost samostojnega gibanja in potrebujejo invalidski voziček. Sposobnost stati se v povprečju ohrani do 16 let.
Ločeno je treba povedati o vpletenosti dihalnih mišic v patološki proces. To opazimo po adolescenci. Slabost diafragme in drugih mišic, ki sodelujejo pri dihanju, vodi do postopnega zmanjšanja zmogljivosti pljuč in volumna prezračevanja. Ponoči je to še posebej opazno (pojavijo se napadi zadušitve), zato imajo otroci lahko strah pred spanjem. Nastane odpoved dihanja, kar poslabša potek sočasnih okužb.
Skeletne deformacije

To so simptomi, povezani s spremembami mišic. Pri otrocih se postopoma oblikujejo povečanje ledvenega upogiba (lordoza), ukrivljenost torakalne hrbtenice v stran (skolioza) in naklon (kifoza), oblika stopala se spremeni. Sčasoma se razvije difuzna osteoporoza. Ti simptomi dodatno prispevajo k poslabšanju gibalnih motenj.
Poškodbe srčne mišice
Je obvezen simptom Duchennove mišične distrofije. Pri bolnikih se razvije kardiomiopatija (hipertrofična ali razširjena). Klinično se to kaže kot motnje srčnega ritma, spremembe krvnega tlaka. Meje srca se povečajo, vendar ima tako veliko srce malo funkcionalnosti. Sčasoma se razvije srčno popuščanje. Kombinacija hudega srčnega popuščanja z motnjami dihanja v ozadju pridružene okužbe je lahko vzrok smrti pri bolnikih z Duchennovo mišično distrofijo.
duševna prizadetost
To ni obvezno, ampak možen znak bolezni. Povezan je s pomanjkanjem posebne oblike distrofina - apodistrofina, ki ga vsebujejo možgani. Okvare inteligence segajo od manjših do idiotskih. Hkrati pa resnost duševnih motenj nikakor ni povezana s stopnjo mišičnih motenj. Socialna neprilagojenost zaradi nezmožnosti prostega gibanja in obiskovanja otroških ustanov (vrtcev, šol) prispeva k poslabšanju kognitivnih motenj.
endokrine motnje
Pojavijo se pri 30-50% bolnikov. Lahko so precej raznoliki, najpogosteje pa gre za debelost s prevladujočim odlaganjem maščobe v mlečnih žlezah, stegnih, zadnjici, ramenskem obroču, nerazvitost (ali disfunkcijo) spolnih organov. Bolniki so pogosto nizke rasti.
Duchennova mišična distrofija vztrajno napreduje. Do starosti 15-20 let skoraj vsi bolniki zaradi nepremičnosti ne morejo skrbeti zase. Na koncu se pridružijo bakterijske okužbe (dihalnih in sečil, okužene preležanine z nezadostno oskrbo), ki v ozadju srčne in dihalne odpovedi vodijo v smrt. Le malo bolnikov preživi mejnik 30 let.
Diagnostika

Diagnoza Duchennove mišične distrofije temelji na več vrstah študij, med katerimi je glavni genetski test (DNK diagnostika).
Samo odkrivanje okvare kromosoma X na območju, ki je odgovorno za sintezo distrofina, zanesljivo potrdi diagnozo. Pred takšno analizo je diagnoza preliminarna.
Druge raziskovalne metode lahko vključujejo:
- določanje aktivnosti kreatin fosfokinaze (CPK). Ta encim odraža smrt mišičnih vlaken. Njegova koncentracija pri Duchennovi mišični distrofiji presega normo za desetine in stokrat do starosti 5 let. Kasneje se raven encima postopoma znižuje, ker so nekatera mišična vlakna že nepovratno uničena;
- elektromiografija. Ta metoda vam omogoča, da potrdite dejstvo, da bolezen temelji na primarnih spremembah mišic, medtem ko so živčni prevodniki popolnoma nedotaknjeni;
- mišična biopsija. Z njegovo pomočjo se določi vsebnost proteina distrofina v mišici. Vendar je ta travmatični postopek v povezavi z izboljšanjem genetske diagnostike v zadnjih desetletjih odšel v ozadje;
- respiratorne preiskave (študija pljučne kapacitete), EKG, ultrazvok srca. Te metode se ne uporabljajo za postavitev diagnoze, so pa potrebne za odkrivanje patoloških sprememb v dihalnem in srčno-žilnem sistemu za odpravo obstoječih motenj.
Identifikacija bolnega otroka v družini pomeni, da ima materin genotip nenormalen X kromosom. V redkih primerih je lahko mati zdrava, če se mutacija pri otroku zgodi po nesreči. Okvarjen kromosom X predstavlja tveganje za prihodnjo nosečnost. Zato bi se moral s takšnimi družinami posvetovati z genetikom. Ob ponavljajočih se nosečnostih staršem ponudimo prenatalno diagnozo, to je študijo genotipa nerojenega otroka, da bi izključili dedne bolezni, vključno z Duchennovo mišično distrofijo.
Za raziskavo boste potrebovali fetalne celice, ki jih pridobimo z različnimi postopki v različnih fazah nosečnosti (na primer biopsija horiona, amniocenteza in drugo). In čeprav te medicinske manipulacije predstavljajo določeno tveganje za nosečnost, vam omogočajo, da natančno odgovorite na vprašanje: ali ima plod genetsko bolezen.
Zdravljenje
Duchennova mišična distrofija je trenutno neozdravljiva bolezen. Otroku (odraslemu) lahko pomagate podaljšati čas telesne aktivnosti z različnimi metodami za ohranjanje mišične moči, kompenzacijo sprememb v srčno-žilnem in dihalnem sistemu.
Kljub temu so napovedi znanstvenikov za popolno ozdravitev te bolezni precej optimistične, saj so prvi koraki v tej smeri že narejeni.
Trenutno se za zdravljenje Duchennove mišične distrofije uporabljajo zdravila:
- steroidi (ob redni uporabi lahko zmanjšajo mišično oslabelost);
- β-2-agonisti (tudi začasno dajejo mišično moč, vendar ne upočasnjujejo napredovanja bolezni).
Uporaba β-2-agonistov (Albuterol, Formoterol) nima statistično pomembnega priznanja, saj je malo izkušenj z njihovo uporabo pri tej patologiji. Kontrolo sprememb zdravstvenega stanja v skupini bolnikov, ki so jemali ta zdravila, smo izvajali eno leto. Zato ni mogoče trditi, da delujejo dlje časa.
Steroidi so danes temelj zdravljenja. Menijo, da vam njihova uporaba omogoča, da nekaj časa ohranite mišično moč, torej lahko upočasnijo napredovanje bolezni. Poleg tega se je izkazalo, da steroidi zmanjšajo tveganje za skoliozo pri Duchennovi mišični distrofiji. Toda kljub temu so možnosti teh zdravil omejene in bolezen bo postopoma napredovala.
 Kdaj se začne hormonsko zdravljenje? Menijo, da je optimalen čas za začetek terapije faza bolezni, ko se motorične sposobnosti ne izboljšajo, vendar se še ne poslabšajo. To se običajno zgodi v starosti 4-6 let. Najpogosteje uporabljena zdravila sta Prednizolon in Deflazacort. Odmerki so predpisani posamezno. Zdravila se uporabljajo, dokler je viden klinični učinek. Ko se začne faza napredovanja bolezni, potem potreba po uporabi steroidov izgine in jih postopoma (!) prekličemo.
Kdaj se začne hormonsko zdravljenje? Menijo, da je optimalen čas za začetek terapije faza bolezni, ko se motorične sposobnosti ne izboljšajo, vendar se še ne poslabšajo. To se običajno zgodi v starosti 4-6 let. Najpogosteje uporabljena zdravila sta Prednizolon in Deflazacort. Odmerki so predpisani posamezno. Zdravila se uporabljajo, dokler je viden klinični učinek. Ko se začne faza napredovanja bolezni, potem potreba po uporabi steroidov izgine in jih postopoma (!) prekličemo.
Od zdravil se za Duchennovo mišično distrofijo uporabljajo tudi srčna zdravila (antiaritmična, presnovna, zaviralci angiotenzinske konvertaze). Omogočajo vam, da se ukvarjate s kardiološkimi vidiki bolezni.
Od metod zdravljenja brez zdravil igrata pomembno vlogo fizioterapija in ortopedska oskrba. Fizioterapevtske tehnike vam omogočajo, da ohranite prožnost in gibljivost sklepov dlje kot brez njihove uporabe, ohranite mišično moč. Dokazano je, da zmerna telesna aktivnost pozitivno vpliva na potek bolezni, nedejavnost in počitek v postelji pa nasprotno prispevata k še hitrejšemu napredovanju bolezni. Zato je treba vzdrževati izvedljivo telesno aktivnost čim dlje, tudi potem, ko je bolnik »preselil« na invalidski voziček. Prikazani so redni tečaji masaže. Plavanje pozitivno vpliva na počutje bolnika.
Ortopedski pripomočki lahko močno olajšajo življenje bolnika. Njihov seznam je precej širok in raznolik: to so različne vrste vertikalizatorjev (pomagajo vzdrževati stoječi položaj), in naprave za samostoječe, in invalidski vozički z električnim pogonom ter posebne pnevmatike za odpravo kontraktur v spodnjem delu noge (uporabljene tudi ponoči), pa stezniki za hrbtenico in dolge opornice za noge (ortoze za koleno-gleženj) in še marsikaj.
Kadar bolezen prizadene tudi dihalne mišice in spontano dihanje postane neučinkovito, je možna uporaba naprav za umetno prezračevanje pljuč različnih modifikacij.
In vendar tudi uporaba vseh teh ukrepov v kompleksu ne omogoča premagovanja bolezni. Do danes obstaja številna obetavna področja raziskav, ki so lahko preboj pri zdravljenju Duchennove mišične distrofije. Najpogostejši med njimi so:
- genska terapija (uvedba "pravilnega" gena z uporabo virusnih delcev, dostava genetskih konstruktov v sestavi liposomov, oligopeptidov, polimernih nosilcev in drugih);
- regeneracija mišičnih vlaken s pomočjo matičnih celic;
- presaditev miogenih celic, ki so sposobne sintetizirati normalni distrofin;
- preskakovanje eksona (z uporabo protismiselnih oligoribonukleotidov) kot poskus upočasnitve napredovanja bolezni in ublažitve njenega poteka;
- zamenjava distrofina z drugim proteinskim utrofinom, katerega gen je dekodiran. Tehnika je bila testirana na miših s pozitivnimi rezultati.
Vsak od novih dogodkov prinaša upanje bolnikom z Duchennovo mišično distrofijo za popolno okrevanje.
Tako je Duchennova mišična distrofija genetski problem pri moških. Za bolezen je značilna progresivna mišična oslabelost zaradi uničenja mišičnih vlaken. Trenutno je neozdravljiva bolezen, vendar si mnogi znanstveniki po vsem svetu prizadevajo ustvariti radikalen način za boj proti njej.
Animirani film Duchennova mišična distrofija glasovno igranje, podnapisi v ruščini:
Mišična distrofija je skupina patologij kronične dedne narave, za katere je značilen progresiven potek, pa tudi trajne histološke motnje.
Sodobne raziskovalne metode v smislu molekularne genetike aktivno širijo razumevanje in razumevanje velikega števila vrst distrofije. Najpomembnejše med njimi so mišične oblike Beckerjeve in Duchennove distrofije ter stanja, ki so po avtosomno prevladujočem tipu dedna progresivna oftalmološka oblika mišične distrofije.
Do zdaj ni bilo izumljenih sredstev, ki bi pomagala popolnoma znebiti distrofije v mišicah.
Obstajajo štiri oblike te patologije. Najpogosteje se postavi diagnoza Duchennove mišične distrofije - v polovici vseh primerov patologije. Ponavadi. Potek bolezni se začne že v otroštvu in povzroči smrt do dvajsetega leta. Beckerjeva mišična distrofija napreduje nekoliko počasneje, bolniki živijo do štiridesetega leta starosti. Druge oblike bolezni običajno ne vplivajo na trajanje človeškega življenja.
Etiološki dejavniki, ki povzročajo distrofijo v mišicah
Nastanek distrofije v mišicah je posledica vpliva različnih genov. Patologijo Duchenne in Becker povzročajo geni, ki se nahajajo na spolnih kromosomih. Te oblike so značilne samo za moške. Druge lezije niso v korelaciji s spolnimi kromosomi, zato lahko prizadenejo tako moške kot ženske.
Glavne manifestacije in znaki napredovanja bolezni
Vse vrste mišične distrofije izzovejo aktiven razvoj mišične atrofije, vendar se lahko razlikujejo glede na resnost patologije in čas njenega nastanka.
- Duchennova distrofija se kaže že v zgodnjem otroštvu - približno med tremi in petimi leti. Ob tem bolniki hodijo v razbitini, težko se vzpenjajo po stopnicah, pogosto postrežejo kar iz jasnega in ne morejo teči. Ko otrok s takšno diagnozo dvigne roke, se zdi, da se njegove lopatice odmaknejo od telesa. Otrok s to vrsto distrofije je priklenjen na invalidski voziček pri starosti 10-12 let, nenehno progresivno oslabitev mišic pa povzroči smrt zaradi nenadnega srčnega popuščanja, odpovedi dihanja ali infekcijskih lezij.
- Beckerjeva distrofija ima veliko podobnosti s prejšnjo vrsto patologije, vendar napreduje veliko počasneje. Simptomi mišične distrofije se začnejo pojavljati šele pri petem letu starosti, po petnajstih letih pa lahko bolniki še vedno samostojno hodijo, včasih celo veliko dlje.
- Ramensko-obrazna oblika mišične distrofije napreduje zelo počasi, njen potek je relativno benigen. V bistvu se bolezen občuti pri starosti 10 let, lahko pa se pokaže tudi v začetku adolescence. Otroci s to diagnozo že v otroštvu slabo sesajo, v starejši starosti pa ne morejo zložiti ustnic v cev, dvigniti roke nad glavo. Za obraz je značilna slaba mimika med jokom ali smehom, vendar je mimika na trenutke še vedno prisotna, kljub temu pa se zelo razlikuje od običajne.
V zdravstvenih centrih, ki so opremljeni z najsodobnejšo tehnologijo za izvajanje imunoloških in molekularnih preiskav, lahko specialisti natančno ugotovijo, ali bo otrok v prihodnosti zbolel za mišično distrofijo. V takih ustanovah organizirajo tudi preglede za starše in sorodnike otroka in razkrijejo prisotnost genov v njih, ki določajo nastanek Beckerjeve ali Duchennove mišične distrofije.
Kako poteka proces zdravljenja?
V sodobni medicini še niso bile razvite metode za preprečevanje ali napredovanje aktivnega razvoja te patologije. Zdravljenje mišične distrofije vključuje organizacijo nasprotovanja zapletom, na primer deformacijam vretenc zaradi šibkih hrbtnih mišic, nagnjenosti telesa k okužbi s pljučnico zaradi oslabitve dihalnih mišic.
Bolnikom z dodatnim razvojem srčnega bloka se lahko izvede implantacija srčnega spodbujevalnika. Za zdravljenje srčnih lezij se priporoča zdravilo fenigidin. Sprejem različnih ortopedskih vam omogoča, da okrepite viseče noge, obnovite delovanje gleženj in zmanjšate pogostost padcev.
Pravilno izbrano usposabljanje pozitivno vpliva tudi na potek patologije. Z razvojem atrofije se za zdravljenje in obnovitveno terapijo uporabljajo anabolične skupine steroidov. Z močno manifestacijo miotoničnih simptomov lezije je predpisan potek zdravljenja z difeninom, ki traja dva do tri tedne. Prav difenin verjetno zavira patološki učinek na sinaptično prevodnost in zmanjša tudi posttetanično mišično aktivnost. Zdravilo Selegin daje pozitivne rezultate, če ga jemljete za popravljanje vzorcev spanja in odpravo visoke zaspanosti. 
Učinkovito zdravljenje je mogoče izvesti le zahvaljujoč genski terapiji, ki se trenutno aktivno razvija. Veliko eksperimentalnih del kaže na izboljšanje stanja mišičnih vlaken pri zdravljenju določenih oblik bolezni. Z razvojem Beckerjeve in Duchennove distrofije pride do nezadostne proizvodnje mišičnih beljakovin - distrofina. Gen, ki je odgovoren za nastanek tega proteina, je največji gen med vsemi znanimi v medicini, v zvezi s čimer so znanstveniki poustvarili mini različico tega gena, adenovirusi pa so postali najboljši prevodniki gena v mišice.
Mišična distrofija je skupina dednih bolezni, pri katerih se mišična masa in njena funkcija postopoma zmanjšujeta.
Sorte mišične distrofije
Devet vrst mišične distrofije vključuje:
Duchennova mišična distrofija (DMD) , ki prizadene dečke tako, da povzroči progresivno mišično oslabelost in se običajno začne v nogah. To je najhujša oblika mišične distrofije.
Beckerjeva mišična distrofija (BMD) , ki prizadene starejše dečke in mlade moške, je blažji od DMD.
Emery-Dreyfusova mišična distrofija (EDMD) , ki prizadene dečke, povzroča kontrakture in oslabelost teleta, oslabelost ramen in nadlakti ter okvare prevodnosti srca. Ženske z EDMD so ogrožene zaradi srčnega bloka.
Mišična distrofija okončin (LGMD) , ki se začne v poznem otroštvu, zgodnji odrasli dobi in prizadene tako moške kot ženske, kar povzroča šibkost v mišicah okoli bokov in ramen. To je najbolj nedosledna oblika mišične distrofije in je razdeljena na več različnih vrst. Mnogim ljudem s sumom na PHMD so v preteklosti verjetno postavili napačno diagnozo; zato je razširjenost bolezni težko oceniti.
Ramensko-skapularno-obrazna miopatija (FSH) , znana tudi kot Landouzy-Dejerine bolezen, ki se začne v poznem otroštvu, zgodnji odrasli dobi in prizadene tako moške kot ženske, kar povzroča šibkost v mišicah obraza, ramen in podlakti. Lahko so prizadeti tudi boki in noge.
miotonična distrofija (MD) , znana tudi kot Steinertova bolezen, ki prizadene tako moške kot ženske, kar ima za posledico splošno šibkost, ki prizadene obraz, noge in roke, in jo spremlja nezmožnost sprostitve prizadetih mišic (miotonija). Simptomi se lahko pojavijo kadar koli od rojstva do odrasle starosti.
Okulofaringealna mišična distrofija (OPMD) , ki prizadene odrasle osebe obeh spolov, kar povzroča oslabelost mišic oči in grla.
Distalna mišična distrofija (DD) , ki se začne v srednjih letih ali kasneje, kar povzroča šibkost v mišicah rok in nog.
Prirojena mišična distrofija (CMD) , ki je prisoten od rojstva, vodi v splošno šibkost in običajno počasi napreduje. Njegov podtip, imenovan Fukuyama CMD, vključuje tudi duševno zaostalost. Obe bolezni sta izjemno redki.
Vzroki in simptomi mišične distrofije
Nekatere oblike mišične distrofije, vključno z DMD, BMD, CMD in večino oblik PHMD, povzročajo okvare genov kompleksa mišičnih beljakovin. Napake v beljakovinah kompleksa vodijo do mišične distrofije, ki postopoma izčrpa svojo sposobnost samopopravljanja. DMD in BMD sta posledica okvar v genu za beljakovino, imenovano distrofin. Razlike med drugimi boleznimi so manj jasne.
 Mišična distrofija je genetska bolezen, kar pomeni, da jo povzročajo okvare genov. Geni, ki so med seboj povezani na kromosomih, imajo dve funkciji. Kodirajo za proizvodnjo beljakovin in so material dedovanja. Starši svojim otrokom preko genov posredujejo celoten sklop navodil za izdelavo lastnih beljakovin.
Mišična distrofija je genetska bolezen, kar pomeni, da jo povzročajo okvare genov. Geni, ki so med seboj povezani na kromosomih, imajo dve funkciji. Kodirajo za proizvodnjo beljakovin in so material dedovanja. Starši svojim otrokom preko genov posredujejo celoten sklop navodil za izdelavo lastnih beljakovin.
Ker oba starša prenašata genetski material na svojega otroka, otrok nosi dve kopiji vsakega gena, po eno od vsakega starša. Pri nekaterih boleznih morata biti obe kopiji okvarjeni. Takšne bolezni imenujemo avtosomno recesivne. Nekatere oblike LGMD in DD kažejo ta vzorec dedovanja, tako kot CMD. Oseba, ki ima samo eno napačno kopijo, se imenuje nosilec in ne bo imela bolezni, lahko pa napačen gen prenese na svoje otroke.
Druge bolezni se pojavijo, ko je poškodovana samo ena kopija gena. Takšne bolezni se imenujejo avtosomno dominantne. Ta vzorec dedovanja prikazujejo PHMD, DM, FSN, OPMD in nekatere oblike DD.
Za vse vrste mišične distrofije je glavni simptom mišična oslabelost. Porazdelitev simptomov, starost, začetek in napredovanje bolezni se močno razlikujejo. Bolečina je tudi simptom mišične distrofije, običajno zaradi učinkov šibkosti.
Diagnoza mišične distrofije
Diagnoza mišične distrofije vključuje temeljit pregled bolnikove zdravstvene anamneze in temeljit zdravniški pregled. Družinska anamneza lahko zagotovi pomembne namige, saj so vse oblike mišične distrofije genetske.
Laboratorijski diagnostični testi za mišično distrofijo lahko vključujejo naslednje:
 Raven mišičnega encima kreatin kinaze (CK) v krvi.
Raven CK se poveča zaradi poškodbe mišic in se lahko v nekaterih primerih opazi, še preden se pojavijo simptomi.
Raven mišičnega encima kreatin kinaze (CK) v krvi.
Raven CK se poveča zaradi poškodbe mišic in se lahko v nekaterih primerih opazi, še preden se pojavijo simptomi.
mišična biopsija pri katerem se za pregled odstrani majhen košček mišičnega tkiva. Za različne oblike mišične distrofije so značilne spremembe v strukturi mišičnih celic in prisotnost vlaknastega tkiva ali drugih aberantnih struktur. Mišično tkivo lahko pregledamo tudi glede prisotnosti ali odsotnosti specifičnih beljakovin, vključno z distrofinom.
elektromiogram (EMG) . EMG se uporablja za preučevanje odziva mišic na stimulacijo. Zmanjšan odziv se kaže v mišični distrofiji.
Genetski testi . Nekatere vrste mišične distrofije je mogoče identificirati s testiranjem na prisotnost mutiranega gena.
Natančni genetski testi za DMD, BMD, DM, več oblik LGMD in EDMD.
Drugi specifični testi po potrebi. Če obstaja sum na EDMD in BMD, bo morda potreben elektrokardiogram za testiranje delovanja srca.
V primeru večine oblik mišične distrofije natančne diagnoze ni težko postaviti. Vendar pa obstajajo tudi izjeme. Tudi z biopsijo je težko razlikovati med FSH in drugo mišično motnjo, polimiozitisom. Mišično distrofijo pri otrocih z začetnim PHMD pogosto zamenjujemo z veliko bolj splošno DMD, zlasti če se pojavi pri dečkih. Zgodnji pojav BMD je zelo podoben DMD. Mišično distrofijo pri otrocih lahko zamenjamo z eno od bolezni motoričnih nevronov, kot je spinalna mišična atrofija; bolezni živčno-mišičnega spoja (miastenija gravis); in druge mišične bolezni.
Zdravljenje mišične distrofije
 Strogo gledano, ni posebnih zdravil za zdravljenje katere koli vrste mišične distrofije. Prednizon in kortikosteroidi so indicirani za upočasnitev napredovanja DMD do neke mere. Prednizolon je predpisan tudi za BMD.
Strogo gledano, ni posebnih zdravil za zdravljenje katere koli vrste mišične distrofije. Prednizon in kortikosteroidi so indicirani za upočasnitev napredovanja DMD do neke mere. Prednizolon je predpisan tudi za BMD.
Zdravljenje mišične distrofije je primarno usmerjeno v preprečevanje zapletov, vključno z zmanjšano gibljivostjo, spretnostjo, kontrakturami, skoliozo, srčnimi napakami in odpovedjo dihanja.
Fizikalna terapija, zlasti redno raztezanje, se uporablja za ohranjanje obsega gibanja prizadetih mišic in za preprečevanje ali odložitev kontrakture. Naramnice se pogosteje uporabljajo na gležnjih in nogah. Krepitev drugih mišičnih skupin za kompenzacijo šibkosti je lahko možna, če so prizadete mišice majhne in izolirane ali v zgodnjih fazah blažjih vrst mišične distrofije. Redna vadba medtem pomaga ohranjati splošno zdravje. Resna telesna aktivnost na splošno ni priporočljiva.
Ko postanejo kontrakture bolj izrazite, se lahko izvede operacija.
Zanikanje odgovornosti: Informacije v tem članku o mišični distrofiji pri otrocih so namenjene samo obveščanju bralca. Ne more biti nadomestilo za nasvet zdravstvenega delavca.
Priporočamo tudi
 Stikalno napajanje: popravilo in izboljšanje
Stikalno napajanje: popravilo in izboljšanje
 Daljinski nadzor svetlobe
Daljinski nadzor svetlobe
 Učne ure plavanja za predšolske otroke
Učne ure plavanja za predšolske otroke
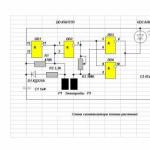 Opombe za mojstra - domači gospodinjski alarmi
Opombe za mojstra - domači gospodinjski alarmi
 Propeler ure na Atmega8
Propeler ure na Atmega8
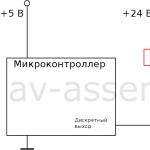 Primeri uporabe naprav in relejev, kako izbrati in pravilno povezati rele Mikrokrmilnik in rele preprosta preklopna vezja
Primeri uporabe naprav in relejev, kako izbrati in pravilno povezati rele Mikrokrmilnik in rele preprosta preklopna vezja