Co to jest hamowanie agregacji płytek krwi? Leki wpływające na hemostazę płytek naczyniowych Agregację płytek krwi hamuje bloker Ca2
Wynalazek dotyczy fizjologii, farmakologii i chemii medycznej, w szczególności ulepszonego sposobu hamowania agregacji płytek krwi. Cel ten osiąga się poprzez opisaną ulepszoną metodę hamowania agregacji płytek krwi za pomocą związków heterocyklicznych zawierających siarkę, która polega na wykorzystaniu znanego benzo-1,2-ditiolo-3-tionu (BDTT) lub jego pochodnych o wzorze ogólnym jako związków zawierających siarkę związki heterocykliczne
 gdzie R1-R4 =H lub C1-4-alkil. Wynik techniczny: umożliwia skuteczniejsze hamowanie agregacji płytek krwi in vitro. Wynalazek dotyczy fizjologii, farmakologii i chemii medycznej, w szczególności ulepszonego sposobu hamowania agregacji płytek krwi. Wyniki licznych badań wskazują, że zwiększona agregacja płytek krwi odgrywa: kluczową rolę w patogenezie wielu chorób układu krążenia (np. zakrzepica tętnicza, zawał mięśnia sercowego, niestabilna dławica piersiowa). Związki mogące zapobiegać aktywacji płytek krwi, która może być wywołana przez endogenne induktory (na przykład adenozyno-5'-difosforan/ADP), są obecnie szeroko stosowane w celu skutecznego oddziaływania na zmiany patologiczne w układzie hemostatycznym (B.A. Sidorenko, D.V. Preobrazhensky. Kliniczne zastosowanie leków przeciwzakrzepowych - M., 1998). Znane są różne metody hamowania agregacji płytek krwi za pomocą związków bicyklicznych zawierających siarkę. Zatem 5-[(2-chlorofenylo)metylo]-4 znalazł praktyczne zastosowanie w zapobieganiu agregacja płytek krwi, 5,6,7-tetrahydrotienopirydyna (tyklopidyna) o wzorze I
gdzie R1-R4 =H lub C1-4-alkil. Wynik techniczny: umożliwia skuteczniejsze hamowanie agregacji płytek krwi in vitro. Wynalazek dotyczy fizjologii, farmakologii i chemii medycznej, w szczególności ulepszonego sposobu hamowania agregacji płytek krwi. Wyniki licznych badań wskazują, że zwiększona agregacja płytek krwi odgrywa: kluczową rolę w patogenezie wielu chorób układu krążenia (np. zakrzepica tętnicza, zawał mięśnia sercowego, niestabilna dławica piersiowa). Związki mogące zapobiegać aktywacji płytek krwi, która może być wywołana przez endogenne induktory (na przykład adenozyno-5'-difosforan/ADP), są obecnie szeroko stosowane w celu skutecznego oddziaływania na zmiany patologiczne w układzie hemostatycznym (B.A. Sidorenko, D.V. Preobrazhensky. Kliniczne zastosowanie leków przeciwzakrzepowych - M., 1998). Znane są różne metody hamowania agregacji płytek krwi za pomocą związków bicyklicznych zawierających siarkę. Zatem 5-[(2-chlorofenylo)metylo]-4 znalazł praktyczne zastosowanie w zapobieganiu agregacja płytek krwi, 5,6,7-tetrahydrotienopirydyna (tyklopidyna) o wzorze I  będący antagonistą płytkowych receptorów P2T ADP (patent USA nr 4963559, kl. 514-301, op. 1990). Wykazano również, że blokerem kanału wapniowego jest D-cis-3-acetoksy-2,3-. chlorowodorek dihydro-5-2-(2-metoksyfenylo)-1,5-benzotiazepino-4(5H)-onu (diltiazem) o wzorze II
będący antagonistą płytkowych receptorów P2T ADP (patent USA nr 4963559, kl. 514-301, op. 1990). Wykazano również, że blokerem kanału wapniowego jest D-cis-3-acetoksy-2,3-. chlorowodorek dihydro-5-2-(2-metoksyfenylo)-1,5-benzotiazepino-4(5H)-onu (diltiazem) o wzorze II  ma działanie przeciwzakrzepowe (A.R.Dehpour, T.Samadian i wsp. „Efekty diltiazemu i werapamilu na zmianę kształtu i agregację płytek królika wywołaną ADP” Gen. Pharmacol, 1995, v.26, No. 6, str.1295-1299 ). Powyższe związki in vitro hamują agregację płytek krwi wywołaną ADP w stężeniu >300 µM Benzo-1,2-ditiolo-3-tion (BDTT) i jego pochodne o wzorze ogólnym III są znane.
ma działanie przeciwzakrzepowe (A.R.Dehpour, T.Samadian i wsp. „Efekty diltiazemu i werapamilu na zmianę kształtu i agregację płytek królika wywołaną ADP” Gen. Pharmacol, 1995, v.26, No. 6, str.1295-1299 ). Powyższe związki in vitro hamują agregację płytek krwi wywołaną ADP w stężeniu >300 µM Benzo-1,2-ditiolo-3-tion (BDTT) i jego pochodne o wzorze ogólnym III są znane.  gdzie R1R4 =H lub C1-6-alkil, benzoil, (podstawiona) grupa aminosulfonylowa itp., jako produkty syntezy organicznej (patent europejski nr 0043936, op. 1981). Nie badano agregacji płytek krwi. Najbardziej zbliżona do opisanej jest znana metoda hamowania agregacji płytek krwi poprzez wprowadzenie do pożywki zawierającej te komórki związków heterocyklicznych zawierających siarkę – podstawionej 3,4-dihydro-1,2-ditiine-2. W szczególności metoda ta doprowadziła do zmniejszenia agregacji płytek krwi przy użyciu 3,4-dihydro-3-winylo-1,2-ditiino-2-tlenku o wzorze IV.
gdzie R1R4 =H lub C1-6-alkil, benzoil, (podstawiona) grupa aminosulfonylowa itp., jako produkty syntezy organicznej (patent europejski nr 0043936, op. 1981). Nie badano agregacji płytek krwi. Najbardziej zbliżona do opisanej jest znana metoda hamowania agregacji płytek krwi poprzez wprowadzenie do pożywki zawierającej te komórki związków heterocyklicznych zawierających siarkę – podstawionej 3,4-dihydro-1,2-ditiine-2. W szczególności metoda ta doprowadziła do zmniejszenia agregacji płytek krwi przy użyciu 3,4-dihydro-3-winylo-1,2-ditiino-2-tlenku o wzorze IV.  przy stosunkowo wysokich stężeniach (np. w warunkach in vitro dla danego związku, IC50 = 100 µM) (patent japoński 02204487, C 07 D 339/08, op. 1991, Chem. Abstr., 1991, t. 114, str. 6523s - prototyp). Celem wynalazku jest opracowanie skuteczniejszej metody hamowania agregacji płytek krwi, cel ten osiąga się opisaną metodą hamowania agregacji płytek krwi poprzez wprowadzenie do pożywki zawierającej te komórki związków heterocyklicznych zawierających siarkę, znamienny tym, że znany BDTT lub jego pochodne o powyższym wzorze ogólnym III, w którym R1-R4 mają podane wartości. Związki o wzorze ogólnym III otrzymano w znany sposób - w reakcji odpowiedniego niepodstawionego lub podstawionego o-chloro - lub chlorek lub bromek o-bromobenzylu z siarką w temperaturze 50-200°C w środowisku eter monoalkilowy glikolu w obecności alkoholanu metalu alkalicznego. Opisany wynalazek ilustrują następujące przykłady. Przykład 1. Hamowanie ludzkich płytek krwi agregację pod wpływem BDTT i jego pochodnych. Wpływ produktów według niniejszego wynalazku na agregację ludzkich płytek krwi badano dobrze znaną metodą turbidymetryczną Borna. W tym celu krew żylną pobraną o godzinie 8 rano od zdrowych dawców wirowano przy 450 g w temperaturze pokojowej w plastikowym pojemniku przez 10 minut, stosując cytrynian sodu jako antykoagulant. Zebrano supernatant, czyli osocze bogate w płytki krwi, i odwirowano przy 650 g przez 30 minut, aby otrzymać osocze ubogie w płytki krwi. Stężenie płytek krwi w osoczu bogatopłytkowym doprowadzono do 2,5108 komórek/ml przez rozcieńczenie osoczem ubogim w płytki krwi i otrzymaną zawiesinę przelano do kuwety o pojemności 0,5 ml. Agregację indukowano przez dodanie ADP do stężenia 20 µM. W każdym doświadczeniu dobierano stężenie ADP tak, aby agregacja była odwracalna, a maksimum występowało po 2 minutach od dodania ADP, nie przekraczając 50%. Rozpraszanie światła w zawiesinie płytek krwi mierzono za pomocą agregometru opracowanego na Wydziale Biologii Uniwersytetu Moskiewskiego. M.V. Łomonosow. Badany związek (w razie potrzeby w postaci roztworu wodnego zawierającego DMSO do 0,2%) dodano do próbki przed dodaniem induktora agregacji (ADP). Produkty według niniejszego wynalazku w stężeniu 1-100 µM hamowało agregację płytek krwi wywołaną 20 µM ADP, natomiast skuteczne działanie zaobserwowano przy stężeniu<30 мкМ. В частности, значение концентрации, при которой достигалось полумаксимальное ингибирование, (IC 50) для БДТТ составляло 6,4 мкМ. Согласно данным прототипа, для известного аналога (3,4-дигидро-3-винил-1,2-дитиин-2-оксида) в условиях in vitro IC 50 имела значение 100 мкМ.Пример 2. Острая токсичность БДТТ и его производных.Острую токсичность соединений настоящего изобретения определяли известным способом по ЛД 50 с использованием беспородных мышей обоего пола средней массой 21 г при комнатной температуре; стандартное питание и воду давали ad libitum в течение всего эксперимента; подвижность животных не ограничивали. Растворы соединений в ДМСО вводили с помощью стерильного шприца внутрибрюшинно. После инъекции за животными вели наблюдение в течение 48 ч по истечении этого времени за мышами наблюдали дополнительно в течение 72 ч (ни одно из животных не погибло в течение дополнительного промежутка времени). Полученные результаты свидетельствуют о том, что значение ЛД 50 для БДТТ составляет >100 mg/kg, a dla jego pochodnych nie mniej niż 50 mg/kg Z danych przedstawionych w przykładzie 1 wynika, że opisana metoda umożliwia skuteczniejsze hamowanie agregacji płytek krwi in vitro w porównaniu do metody znanej.
przy stosunkowo wysokich stężeniach (np. w warunkach in vitro dla danego związku, IC50 = 100 µM) (patent japoński 02204487, C 07 D 339/08, op. 1991, Chem. Abstr., 1991, t. 114, str. 6523s - prototyp). Celem wynalazku jest opracowanie skuteczniejszej metody hamowania agregacji płytek krwi, cel ten osiąga się opisaną metodą hamowania agregacji płytek krwi poprzez wprowadzenie do pożywki zawierającej te komórki związków heterocyklicznych zawierających siarkę, znamienny tym, że znany BDTT lub jego pochodne o powyższym wzorze ogólnym III, w którym R1-R4 mają podane wartości. Związki o wzorze ogólnym III otrzymano w znany sposób - w reakcji odpowiedniego niepodstawionego lub podstawionego o-chloro - lub chlorek lub bromek o-bromobenzylu z siarką w temperaturze 50-200°C w środowisku eter monoalkilowy glikolu w obecności alkoholanu metalu alkalicznego. Opisany wynalazek ilustrują następujące przykłady. Przykład 1. Hamowanie ludzkich płytek krwi agregację pod wpływem BDTT i jego pochodnych. Wpływ produktów według niniejszego wynalazku na agregację ludzkich płytek krwi badano dobrze znaną metodą turbidymetryczną Borna. W tym celu krew żylną pobraną o godzinie 8 rano od zdrowych dawców wirowano przy 450 g w temperaturze pokojowej w plastikowym pojemniku przez 10 minut, stosując cytrynian sodu jako antykoagulant. Zebrano supernatant, czyli osocze bogate w płytki krwi, i odwirowano przy 650 g przez 30 minut, aby otrzymać osocze ubogie w płytki krwi. Stężenie płytek krwi w osoczu bogatopłytkowym doprowadzono do 2,5108 komórek/ml przez rozcieńczenie osoczem ubogim w płytki krwi i otrzymaną zawiesinę przelano do kuwety o pojemności 0,5 ml. Agregację indukowano przez dodanie ADP do stężenia 20 µM. W każdym doświadczeniu dobierano stężenie ADP tak, aby agregacja była odwracalna, a maksimum występowało po 2 minutach od dodania ADP, nie przekraczając 50%. Rozpraszanie światła w zawiesinie płytek krwi mierzono za pomocą agregometru opracowanego na Wydziale Biologii Uniwersytetu Moskiewskiego. M.V. Łomonosow. Badany związek (w razie potrzeby w postaci roztworu wodnego zawierającego DMSO do 0,2%) dodano do próbki przed dodaniem induktora agregacji (ADP). Produkty według niniejszego wynalazku w stężeniu 1-100 µM hamowało agregację płytek krwi wywołaną 20 µM ADP, natomiast skuteczne działanie zaobserwowano przy stężeniu<30 мкМ. В частности, значение концентрации, при которой достигалось полумаксимальное ингибирование, (IC 50) для БДТТ составляло 6,4 мкМ. Согласно данным прототипа, для известного аналога (3,4-дигидро-3-винил-1,2-дитиин-2-оксида) в условиях in vitro IC 50 имела значение 100 мкМ.Пример 2. Острая токсичность БДТТ и его производных.Острую токсичность соединений настоящего изобретения определяли известным способом по ЛД 50 с использованием беспородных мышей обоего пола средней массой 21 г при комнатной температуре; стандартное питание и воду давали ad libitum в течение всего эксперимента; подвижность животных не ограничивали. Растворы соединений в ДМСО вводили с помощью стерильного шприца внутрибрюшинно. После инъекции за животными вели наблюдение в течение 48 ч по истечении этого времени за мышами наблюдали дополнительно в течение 72 ч (ни одно из животных не погибло в течение дополнительного промежутка времени). Полученные результаты свидетельствуют о том, что значение ЛД 50 для БДТТ составляет >100 mg/kg, a dla jego pochodnych nie mniej niż 50 mg/kg Z danych przedstawionych w przykładzie 1 wynika, że opisana metoda umożliwia skuteczniejsze hamowanie agregacji płytek krwi in vitro w porównaniu do metody znanej.
Prawo
Sposób hamowania agregacji płytek krwi poprzez wprowadzenie związków heterocyklicznych zawierających siarkę do pożywki zawierającej te komórki, znamienny tym, że jako związki heterocykliczne zawierające siarkę stosuje się benzo-1,2-ditiolo-3-tion i jego pochodne o wzorze ogólnym  gdzie R1-R4 oznacza H lub C1-4 oznacza alkil.
gdzie R1-R4 oznacza H lub C1-4 oznacza alkil.
Podobne patenty:
Wynalazek dotyczy chemii organicznej i farmakologii, a mianowicie mieszaniny izomerów soli potasowej kwasu 2-octowego w stosunku molowym 1:3, wykazującej działanie kardiotoniczne
Układ hemostazy pełni w organizmie następujące główne funkcje:
Utrzymuje krew w naczyniach w stanie płynnym, co jest niezbędne do prawidłowego dopływu krwi do narządów i tkanek;
Zapewnia zatrzymanie krwawienia w przypadku uszkodzenia ściany naczynia.
Zatrzymanie krwawienia (hemostaza - z greckiego hemo- krew, zastój- stop) osiąga się przy udziale kilku mechanizmów. Po uszkodzeniu ściany naczynia następuje skurcz naczyń. Ta natychmiastowa reakcja na uraz może zatrzymać krwawienie tylko wtedy, gdy obrażenia małych naczyń są niewielkie. Zasadniczo zatrzymanie krwawienia osiąga się poprzez tworzenie się skrzepów krwi, które zapobiegają utracie krwi poprzez zamknięcie miejsca urazu. Takie miejscowe tworzenie się skrzepów krwi (czopów hemostatycznych) w przypadku uszkodzenia naczyń jest reakcją ochronną.
Jednakże w pewnych warunkach wewnątrz naczyń tworzą się skrzepy krwi, zamykające ich światło i uniemożliwiające prawidłowy przepływ krwi. Do powstania skrzepliny wewnątrznaczyniowej może dojść przy zmianach patologicznych w śródbłonku naczyń, w tym jego uszkodzeniu związanym z miażdżycą, podwyższonym ciśnieniem krwi lub innymi czynnikami. Przyczyną powstawania zakrzepów mogą być również nieprawidłowe zmiany w przepływie krwi (na przykład zmniejszenie jego prędkości) lub niedobór niektórych białek zapobiegających powstawaniu zakrzepów.
Zakrzepica zachodzi przy udziale dwóch głównych procesów: agregacji płytek krwi i krzepnięcia krwi (hemokoagulacji).
Agregacja płytek krwi - jest to połączenie płytek krwi w konglomeraty (agregaty) o różnych rozmiarach i gęstościach. Proces ten inicjowany jest w momencie uszkodzenia ściany naczynia. W miejscu uszkodzenia płytki krwi najpierw wiążą się z czynnikiem von Willebranda i kolagenem warstwy podśródbłonkowej (występuje adhezja płytek krwi). Interakcja z kolagenem powoduje aktywację płytek krwi (ryc. 27-1). W tym przypadku same płytki stają się źródłem substancji stymulujących agregację, takich jak tromboksan A2, ADP,
serotonina. Trombina, która powstaje lokalnie podczas krzepnięcia krwi, również indukuje agregację płytek krwi. Ponadto katecholaminy, czynnik aktywujący płytki krwi i niektóre inne substancje endogenne są induktorami agregacji.
Agregacji płytek krwi zapobiega prostacyklina i śródbłonkowy czynnik rozkurczający, które są wytwarzane przez komórki śródbłonka naczyń i uwalniane do krwioobiegu. W przypadku uszkodzenia komórek śródbłonka synteza tych substancji maleje i na tym tle dominuje działanie substancji stymulujących agregację. W rezultacie płytki krwi łączą się w agregaty, z których powstaje skrzeplina płytkowa.
Skrzep płytkowy wzmacnia się dzięki powstającym w tym procesie niciom fibrynowym krzepnięcie krwi. Głównymi uczestnikami tego procesu są białka osocza krwi zwane czynnikami krzepnięcia krwi.
Czynniki krzepnięcia osocza są syntetyzowane w wątrobie i krążą we krwi w postaci nieaktywnej. W przypadku uszkodzenia ściany naczynia, przy jego udziale następuje szybka aktywacja czynnika VII czynnik tkankowy- białko przezbłonowe syntetyzowane przez różne komórki (w tym aktywowane komórki śródbłonka) i zwykle nie mające kontaktu z krwią. Ekspresja czynnika tkankowego na powierzchni komórki podczas uszkodzenia śródbłonka znacznie przyspiesza aktywację czynnika VII (jego przemianę w czynnik VIIa) w obecności jonów Ca 2+. Pod wpływem czynnika VIIa (w połączeniu z czynnikiem tkankowym) następuje sekwencyjna aktywacja pozostałych czynników krzepnięcia krwi (IX i X) w złożonym układzie autokatalitycznym zwanym kaskadą krzepnięcia krwi. W efekcie pod wpływem czynnika Xa powstaje trombina (czynnik Ha), która przekształca krążące we krwi rozpuszczalne białko fibrynogen (czynnik I) w nierozpuszczalną fibrynę (ryc. 27-5). Fibryna polimeryzuje i wypełniając przestrzeń między płytkami krwi, wzmacnia skrzeplinę płytkową. Nici fibrynowe przenikają do skrzepu krwi, tworząc sieć, która zatrzymuje czerwone krwinki krążące we krwi. Tworzy się czerwony skrzep krwi.
Krzepnięciu krwi przeciwdziałają substancje będące naturalnymi inhibitorami aktywnych czynników krzepnięcia krwi.
Zapobiega się aktywacji czynnika X przez czynnik VIIa inhibitor szlaku czynników tkankowych, syntetyzowany przez śródbłonek
komórki. Inhibitorem trombiny i niektórych innych aktywnych czynników krzepnięcia (Xa, IXa, XIa, XIIa) jest antytrombina III- białko krążące w osoczu krwi, które działa w połączeniu z heparyną lub substancjami heparynopodobnymi (obecnymi na powierzchni nienaruszonych komórek śródbłonka). Substancje te znacznie przyspieszają inaktywację czynników krzepnięcia krwi pod wpływem antytrombiny III.
Inhibitor czynników VIIIa i Va niezbędnych do tworzenia trombiny - aktywowane białko C. Białko to jest syntetyzowane w wątrobie przy udziale witaminy K, krąży we krwi w postaci nieaktywnej i jest aktywowane przez trombinę na powierzchni nienaruszonych komórek śródbłonka. Aktywacja białka C wzrasta wraz z nadmiernym wytwarzaniem trombiny. Kiedy w miejscu uszkodzenia ściany naczynia tworzą się lokalne skrzepy krwi, powyższe inhibitory pomagają utrzymać krew w stanie ciekłym, zapobiegając wzrostowi skrzepu krwi wewnątrz naczynia.
Agregacja płytek krwi i krzepnięcie krwi są ze sobą powiązane. Przewaga tego lub innego procesu w mechanizmie tworzenia skrzepliny zależy od kalibru naczynia i prędkości przepływu krwi. Agregacja płytek krwi ma większe znaczenie dla tworzenia się skrzepów krwi przy dużych prędkościach przepływu krwi, tj. w tętnicach. W naczyniach żylnych, gdzie prędkość przepływu krwi jest mała, dominuje proces krzepnięcia krwi.
Dalszy los skrzepliny zależy od aktywności układu fibrynolitycznego. Jeżeli ten układ funkcjonuje prawidłowo, fibryna ulega stopniowemu rozpuszczaniu (fibrynoliza) przy udziale enzymu plazminy, który pod wpływem aktywatorów powstaje z nieaktywnego prekursora (plazminogenu). Działaniu plazminy zapobiegają krążące we krwi antyplazminy. Aktywatory plazminogenu są neutralizowane przez specyficzne inhibitory.
Zakłócenie procesów agregacji płytek krwi i krzepnięcia krwi i/lub wzmożona aktywność układu fibrynolitycznego może prowadzić do krwawień, a nadmierna aktywacja tych procesów lub zahamowanie fibrynolizy może prowadzić do powstania wewnątrznaczyniowych zakrzepów krwi (zakrzepicy). W wyniku zakrzepicy naczyń tętniczych zmniejsza się dopływ krwi do tkanek i rozwija się ich niedokrwienie. Konsekwencją niedokrwienia jest śmierć komórki (martwica). Zakrzepica może powodować tak poważne powikłania, jak zawał mięśnia sercowego (zakrzepica tętnicy wieńcowej), udar niedokrwienny mózgu (zakrzepica naczyń mózgowych) itp. Zakrzepica żylna może powodować zatorowość płucną.
Aby zapobiec zakrzepicy, stosuje się substancje hamujące agregację płytek krwi i krzepnięcie krwi, zapobiegając w ten sposób tworzeniu się skrzepów krwi. W przypadku zakrzepicy stosuje się również substancje powodujące lizę skrzepów krwi - środki trombolityczne (fibrynolityczne).
Aby zatrzymać krwawienie, stosuje się środki zwiększające krzepliwość krwi i środki hamujące fibrynolizę. Wybór jednego lub drugiego zależy od przyczyny krwawienia.
Praktyczne znaczenie mają następujące grupy środków wpływających na powstawanie skrzeplin.
Środki zmniejszające agregację płytek krwi(leki przeciwpłytkowe).
Leki wpływające na krzepnięcie krwi.
Leki zmniejszające krzepliwość krwi (antykoagulanty).
Leki zwiększające krzepliwość krwi (leki hemostatyczne).
Środki wpływające na fibrynolizę.
Środki fibrynolityczne (trombolityczne).
Środki przeciwfibrynolityczne (inhibitory fibrynolizy).
27.1. LEKI REDUKUJĄCE AGREGACJĘ Płytek krwi (PRZECIWPŁYTKOWE)
Płytki krwi to małe elementy krwi w kształcie krążka, które powstają jako fragmenty megakariocytów szpiku kostnego. Płytki krążą we krwi przez 6-12 dni, po czym są wychwytywane przez makrofagi tkankowe.
Śródbłonek naczyniowy wpływa na aktywność funkcjonalną płytek krwi. Komórki śródbłonka uwalniają do krwiobiegu prostacyklinę (prostaglandynę I 2) i śródbłonkowy czynnik rozkurczający, utożsamiany z tlenkiem azotu - NO. Substancje te zapobiegają agregacji płytek krwi. Ponadto komórki śródbłonka wydzielają substancje, które zmniejszają krzepnięcie krwi i sprzyjają lizie skrzepu. Wszystko to zapewnia właściwości przeciwzakrzepowe nienaruszonego śródbłonka naczyniowego.
W przypadku uszkodzenia śródbłonka naczyniowego, które może być spowodowane różnymi czynnikami (uraz mechaniczny, infekcje, zmiany miażdżycowe w ścianie naczyń, podwyższone ciśnienie krwi itp.), Właściwości przeciwzakrzepowe śródbłonka ulegają osłabieniu, co stwarza warunki do powstawania zakrzepu krwi. Zaburzona zostaje synteza prostacykliny i śródbłonkowego czynnika rozkurczającego, co ułatwia kontakt
płytki krwi z uszkodzoną powierzchnią śródbłonka. Płytki krwi gromadzą się w miejscu uszkodzenia i oddziałują z podśródbłonkiem naczyń: bezpośrednio lub poprzez czynnik von Willebranda (wydzielany przez aktywowane płytki krwi i komórki śródbłonka), wiążą się z kolagenem i innymi białkami podśródbłonka przy udziale specyficznych glikoprotein zlokalizowanych w płytce krwi membrana. Czynnik von Willebranda wiąże się z glikoproteiną Ib, a kolagen wiąże się z glikoproteiną Ia błony płytek krwi (patrz ryc. 27-1). Oddziaływanie kolagenu (a także trombiny, która tworzy się lokalnie w małych ilościach już w początkowej fazie tworzenia skrzepliny) na płytki krwi powoduje zmianę ich stanu – aktywację. Płytki krwi zmieniają swój kształt (z krążkowego stają się rozsiane wieloma wyrostkami – pseudopodiami) i pokrywają uszkodzoną powierzchnię naczynia.
Po aktywacji płytki krwi uwalniają różne substancje biologicznie czynne, które w nieaktywowanych płytkach krwi znajdują się w granulkach (α-granulki, gęste granulki). Gęste granulki są magazynem substancji stymulujących agregację płytek krwi: ADP i serotoniny. Uwalnianie tych substancji z ziarnistości płytek krwi następuje w wyniku wzrostu wewnątrzkomórkowego stężenia Ca 2+ w wyniku działania na płytki krwi kolagenu, trombiny i innych induktorów agregacji, w tym samego ADP. ADP uwalniany do krwioobiegu pobudza specyficzne (purynergiczne) receptory zlokalizowane w błonie płytek krwi. Poprzez receptory sprzężone z białkiem G (receptory purynergiczne P2Y 12) ADP powoduje hamowanie cyklazy adenylanowej i obniżenie poziomu cAMP, co prowadzi do wzrostu poziomu Ca 2 w cytoplazmie płytek krwi (ryc. 27-2).
Ponadto, po aktywacji płytek krwi, wzrasta aktywność fosfolipazy błonowej płytek krwi A2, enzymu biorącego udział w tworzeniu kwasu arachidonowego z fosfolipidów błonowych. W płytkach krwi cykliczne endonadtlenki (prostaglandyny G2/H2) syntetyzowane są najpierw z kwasu arachidonowego pod wpływem cyklooksygenazy, a z nich przy udziale tromboksanezyny-
Tetaza wytwarza tromboksan A2, aktywny stymulator agregacji płytek krwi i środek zwężający naczynia krwionośne. Po uwolnieniu do krwioobiegu tromboksan A2 stymuluje receptory tromboksanu na błonach płytek krwi. W rezultacie poprzez C wiąże się z tymi receptorami Q -białka fosfolipaza C jest aktywowana i tworzenie
Ryż.27-1. Adhezja i agregacja płytek krwi podczas uszkodzenia ściany naczynia: EC – komórka śródbłonka; VW – współczynnik von Willebranda; TxA 2 - tromboksan A 2; PGI 2 – prostacyklina; NIE - śródbłonkowy czynnik rozkurczający; GP - glikoproteiny; GP lb/lla – glikoproteiny lb/lla (od: Katzung B.G. Bazic and Clinical Pharmacology – NY, 2001, z poprawkami)
1,4,5-trifosforan inozytolu, który sprzyja uwalnianiu Ca 2+ z wewnątrzkomórkowego magazynu płytek krwi (rolę magazynu wapnia w płytkach krwi pełni układ gęstych kanalików). Prowadzi to do wzrostu stężenia cytoplazmatycznego Ca 2+ (ryc. 27-2). Tromboksan A 2 powoduje wzrost stężenia Ca 2+ w komórkach mięśni gładkich naczyń, co prowadzi do zwężenia naczyń.

Ryż. 27-2. Mechanizmy działania leków przeciwpłytkowych (kwas acetylosalicylowy, tyklopidyna i epoprostenol): EC – komórka śródbłonka; PL - fosfolipidy błon komórkowych; AA – kwas archidonowy; PLA 2 - fosfolipaza A 2; COX – cyklooksygenaza; TS – syntetaza tromboksanu; PS – syntetaza prostacykliny; PGG 2 /H 2 - cykliczne endonadtlenki; TxA 2 - tromboksan A 2; PGI 2 – prostacyklina; AC – cyklaza adenylanowa; PLS - fosfolipaza C; IP 3 - inozytol-1,4,5-trifosforan
Zatem ADP i tromboksan A 2 zwiększają poziom Ca 2+ w cytoplazmie płytek krwi. Cytoplazmatyczny Ca 2+ powoduje zmianę konformacji glikoprotein IIb/IIIa w błonie płytek krwi, w wyniku czego nabywają one zdolność wiązania fibrynogenu. Jedna cząsteczka fibrynogenu ma dwa miejsca wiązania glikoprotein IIb/IIIa, dzięki czemu może łączyć dwie płytki krwi (ryc. 27-3). Połączenie wielu płytek krwi mostkami fibrynogenu prowadzi do powstania agregatów płytek krwi.
Prostacyklina (prostaglandyna I 2) ma odwrotny wpływ na agregację płytek krwi. Jak tromboksan, prostacyklina
powstaje z cyklicznych endonadtlenków, ale pod wpływem innego enzymu - syntetazy prostacykliny. Prostacyklina jest syntetyzowana przez komórki śródbłonka i uwalniana do krwioobiegu, gdzie poprzez białko Gs stymuluje receptory prostacykliny w błonie płytek krwi i związaną z nimi cyklazę adenylanową. W rezultacie wzrasta poziom cAMP w płytkach krwi i zmniejsza się stężenie cytoplazmatycznego Ca 2+ (patrz ryc. 27-2). Zapobiega to zmianie konformacji glikoprotein IIb/IIIa i utracie przez nie zdolności wiązania fibrynogenu. W ten sposób prostacyklina zapobiega agregacji płytek krwi. Pod wpływem prostacykliny zmniejsza się stężenie Ca 2+ w komórkach mięśni gładkich naczyń, co prowadzi do rozszerzenia naczyń.

Można wyróżnić następującą sekwencję głównych zdarzeń prowadzących do agregacji płytek krwi (patrz Diagram 27-1).
Główny nacisk działania leków przeciwpłytkowych stosowanych obecnie w praktyce klinicznej wiąże się z eliminacją działania tromboksanu A 2 i ADP oraz blokadą glikoprotein IIb/IIIa błon płytek krwi. Stosuje się również substancje o innym mechanizmie działania, które zwiększają stężenie cAMP w płytkach krwi, a tym samym zmniejszają w nich stężenie Ca 2+.
Wyróżnia się następujące grupy środków zmniejszających agregację płytek krwi.
Środki hamujące syntezę tromboksanu A2. - Inhibitory cyklooksygenazy:
kwas acetylosalicylowy.

Schemat 27.1. Mechanizm agregacji płytek krwi
Inhibitory cyklooksygenazy i syntetazy tromboksanu: indobufen.
Środki stymulujące receptory prostacykliny:
epoprostenol**.
Substancje zakłócające działanie ADP na płytki krwi:
tiklopidyna; klopidogrel.
Środki hamujące fosfodiesterazę płytkową:
dipirydamol
Środki blokujące glikoproteiny IIb/IIIa błon płytek krwi.
Przeciwciała monoklonalne: abciximab.
Syntetyczne blokery glikoprotein IIb/IIIa: eptifibatyd; tyrofiban.
Środki hamujące syntezę tromboksanu A 2
Kwas acetylosalicylowy (aspiryna*) jest dobrze znanym środkiem przeciwzapalnym, przeciwbólowym i przeciwgorączkowym. Obecnie szeroko stosowany jako środek przeciwpłytkowy. Działanie przeciwpłytkowe kwasu acetylosalicylowego jest związane z jego hamującym wpływem na syntezę tromboksanu A 2 w płytkach krwi.
Kwas acetylosalicylowy nieodwracalnie hamuje cyklooksygenazę (powoduje nieodwracalną acetylację enzymu), a tym samym zakłóca powstawanie cyklicznych endonadtlenków, prekursorów tromboksanu A2 i prostaglandyn z kwasu arachidonowego. Dlatego pod wpływem kwasu acetylosalicylowego zmniejsza się nie tylko synteza tromboksanu A 2 w płytkach krwi, ale także synteza prostacykliny w komórkach śródbłonka naczyń (patrz ryc. 27-2). Jednakże dobierając odpowiednie dawki i schemat leczenia, można uzyskać preferencyjny wpływ kwasu acetylosalicylowego na syntezę tromboksanu A 2 . Wynika to ze znacznych różnic pomiędzy płytkami krwi i komórkami śródbłonka.
Płytki krwi – komórki pozbawione jądra – nie mają układu resyntezy białek i dlatego nie są w stanie syntetyzować cyklooksygenazy. Zatem przy nieodwracalnym hamowaniu tego enzymu zaburzenie syntezy tromboksanu A2 utrzymuje się przez cały okres życia płytki krwi, tj. w ciągu 7-10 dni. Ze względu na powstawanie nowych płytek krwi działanie przeciwpłytkowe kwasu acetylosalicylowego utrzymuje się krócej, dlatego w celu uzyskania stabilnego działania leku (tj. stabilnego spadku poziomu tromboksanu) zaleca się jego przepisanie raz dziennie.
Cykloksygenaza ulega resyntezie w komórkach śródbłonka naczyń, a aktywność tego enzymu zostaje przywrócona w ciągu kilku godzin po przyjęciu kwasu acetylosalicylowego. Dlatego przepisując lek raz dziennie, nie obserwuje się znaczącego zmniejszenia syntezy prostacykliny.

Ponadto około 30% kwasu acetylosalicylowego ulega metabolizmowi pierwszego przejścia w wątrobie, dlatego jego stężenie w krążeniu ogólnoustrojowym jest niższe niż we krwi wrotnej. W rezultacie kwas acetylosalicylowy działa na płytki krwi krążące w krwiobiegu wrotnym w większym stężeniu niż na komórki śródbłonka naczyń układowych. Dlatego do zahamowania syntezy tromboksanu A2 w płytkach krwi potrzebne są mniejsze dawki kwasu acetylosalicylowego niż do zahamowania syntezy prostacykliny w komórkach śródbłonka.
Z tych powodów wraz ze wzrostem dawki i częstotliwości podawania kwasu acetylosalicylowego nasila się jego hamujący wpływ na syntezę prostacyklin, co może prowadzić do osłabienia działania przeciwpłytkowego. W związku z tymi cechami zaleca się przepisywanie kwasu acetylosalicylowego jako leku przeciwpłytkowego w małych dawkach (średnio 100 mg) raz na dobę.
Jako środek przeciwpłytkowy kwas acetylosalicylowy stosuje się w niestabilnej dławicy piersiowej, w zapobieganiu zawałowi mięśnia sercowego, udarowi niedokrwiennemu i zakrzepicy naczyń obwodowych, aby zapobiec tworzeniu się skrzepów krwi podczas pomostowania aortalno-wieńcowego i angioplastyki wieńcowej. Kwas acetylosalicylowy przepisywany jest doustnie w dawkach 75-160 mg (w niektórych wskazaniach - w zakresie dawek od 50 do 325 mg) raz dziennie przez dłuższy czas. Obecnie lekarze mają do dyspozycji preparaty kwasu acetylosalicylowego przeznaczone do profilaktyki zakrzepicy, które zawierają 50-325 mg substancji czynnej, w tym tabletki dojelitowe – Acecardol*, Aspicor*, Cardiopyrin*, Aspirin Cardio*, Novandol*, Thrombo ACC * itp. Działanie przeciwpłytkowe kwasu acetylosalicylowego następuje szybko (w ciągu 20-30 minut). Postacie dawkowania z powłoką dojelitową zaczynają działać wolniej, ale przy długotrwałym stosowaniu ich skuteczność praktycznie nie różni się od skuteczności konwencjonalnych tabletek. Aby uzyskać szybszy efekt, tabletki kwasu acetylosalicylowego należy żuć.
Główne działania niepożądane kwasu acetylosalicylowego są związane z hamowaniem cyklooksygenazy. Zakłóca to tworzenie prostaglandyn E 2 i I 2, które mają działanie przeciwwydzielnicze i ochronne dla żołądka (zmniejszają wydzielanie kwasu solnego przez komórki okładzinowe żołądka, zwiększają wydzielanie śluzu i wodorowęglanów). W efekcie nawet przy krótkotrwałym stosowaniu kwas acetylosalicylowy może powodować uszkodzenie nabłonka żołądka i dwunastnicy (działanie wrzodujące). Wpływ na błonę śluzową żołądka jest mniej wyraźny w przypadku stosowania postaci dawkowania z powłoką dojelitową. Podczas stosowania kwasu acetylosalicylowego możliwe jest krwawienie z przewodu pokarmowego i inne powikłania krwotoczne. Ryzyko wystąpienia takich powikłań jest mniejsze, gdy kwas acetylosalicylowy przepisywany jest w dawce 100 mg/dobę lub mniejszej. Selektywne hamowanie COX prowadzi do aktywacji szlaku lipooksygenazy do konwersji kwasu arachidonowego i tworzenia leukotrienów, które mają właściwości zwężające oskrzela. U pacjentów z astmą oskrzelową kwas acetylosalicylowy może wywołać atak („astma aspirynowa”). Możliwe są reakcje alergiczne.
Aby zmniejszyć wrzodziejące działanie kwasu acetylosalicylowego, zaproponowano lek złożony Cardiomagnyl* zawierający wodorotlenek magnezu. Wodorotlenek magnezu neutralizuje kwas solny w żołądku (działanie zobojętniające kwas), zmniejszając jego szkodliwy wpływ na błonę śluzową. Lek stosuje się w tych samych wskazaniach co kwas acetylosalicylowy, w tym w profilaktyce wtórnej udaru niedokrwiennego mózgu.
Indobufen (ibustrin*) zmniejsza syntezę tromboksanu A 2, jednocześnie hamując cyklooksygenazę i syntetazę tromboksanu. W przeciwieństwie do kwasu acetylosalicylowego, indobufen powoduje odwracalne hamowanie cyklooksygenazy. Podczas przyjmowania tego leku następuje względny wzrost ilości prostacykliny (wzrasta stosunek prostacykliny do tromboksanu A2). Indobufen hamuje adhezję i agregację płytek krwi. Wskazania do stosowania i skutki uboczne są takie same jak w przypadku kwasu acetylosalicylowego.
Środki stymulujące receptory prostacykliny
Innym sposobem ograniczenia agregacji płytek krwi jest stymulacja receptorów prostacykliny. W tym celu używają
preparat prostacykliny epoprostenol*. Działanie prostacykliny jest odwrotne do działania tromboksanu A 2 nie tylko na płytki krwi, ale także na napięcie naczyniowe. Powoduje rozszerzenie naczyń krwionośnych i obniżenie ciśnienia krwi. To działanie prostacykliny stosuje się w leczeniu nadciśnienia płucnego. Ponieważ prostacyklina jest szybko niszczona we krwi (t 1/2 około 2 minut) i dlatego nie działa długo, lek podaje się w infuzji. Ze względu na krótkie działanie epoprostenol* nie znalazł szerokiego zastosowania jako lek przeciwpłytkowy. Możliwym obszarem zastosowania przeciwpłytkowego działania epoprostenolu jest zapobieganie agregacji płytek krwi podczas krążenia pozaustrojowego.
Środki zakłócające działanie ADP na płytki krwi
Tiklopidyna (tiklid*), pochodna tienopirydyny, hamuje agregację płytek krwi spowodowaną ADP. Tiklopidyna jest prolekiem; jej działanie przeciwpłytkowe wiąże się z tworzeniem aktywnego metabolitu przy udziale mikrosomalnych enzymów wątrobowych. Metabolit tiklopidyny zawiera grupy tiolowe, poprzez które nieodwracalnie wiąże się z receptorami purynergicznymi P2Y12 w błonie płytek krwi. Prowadzi to do eliminacji stymulującego działania ADP na płytki krwi i zmniejszenia w nich stężenia cytoplazmatycznego Ca 2+. W rezultacie zmniejsza się ekspresja glikoprotein IIb/IIIa w błonie płytek krwi i ich wiązanie z fibrynogenem (patrz ryc. 27-2). Ze względu na nieodwracalny charakter działania, tiklopidyna wykazuje długotrwałe działanie przeciwpłytkowe.
Maksymalny efekt przy stałym stosowaniu tiklopidyny osiąga się po 7-11 dniach (czas potrzebny do powstania i rozwoju działania aktywnego metabolitu), a po odstawieniu leku utrzymuje się przez cały czas życia płytek krwi (7-10 dni).
Tyklopidyna jest przepisywana w celu wtórnej profilaktyki udaru niedokrwiennego mózgu, zapobiegania zakrzepicy w chorobach zarostowych kończyn dolnych, podczas pomostowania aortalno-wieńcowego i stentowania tętnic wieńcowych. Lek jest skuteczny po podaniu doustnym, przepisywany 2 razy dziennie podczas posiłków.
Stosowanie tiklopidyny jest ograniczone ze względu na jej skutki uboczne. Możliwa utrata apetytu, nudności, wymioty, biegunka (20%), bóle brzucha, wysypki skórne (11-14%). Odnotowany
zwiększenie poziomu aterogennych lipoprotein w osoczu krwi. Krwawienie jest częstym powikłaniem podczas stosowania leków przeciwpłytkowych. Niebezpiecznym powikłaniem jest neutropenia, która w ciągu pierwszych trzech miesięcy leczenia występuje u 1–2,4% pacjentów. Możliwa jest trombocytopenia, agranulocytoza i bardzo rzadko niedokrwistość aplastyczna. W związku z tym w pierwszych miesiącach leczenia konieczne jest systematyczne monitorowanie obrazu krwi.
Klopidogrel (Plavix*, Zilt*) jest podobny do tiklopidyny pod względem budowy chemicznej, głównych efektów i mechanizmu działania. Podobnie jak tyklopidyna jest prolekiem i ulega przemianie w wątrobie, tworząc aktywny metabolit. Od drugiego dnia kuracji zaobserwowano znaczne zahamowanie agregacji płytek krwi, maksymalny efekt osiągany jest po 4-7 dniach. Po odstawieniu leku jego działanie utrzymuje się przez 7-10 dni. Klopidogrel ma przewagę nad tyklopidyną w działaniu - w dawce dziennej 75 mg powoduje takie samo zmniejszenie agregacji płytek krwi i wydłużenie czasu krwawienia jak tyklopidyna w dawce dziennej 500 mg.

Klopidogrel stosuje się w tych samych wskazaniach co kwas acetylosalicylowy, w przypadku nietolerancji. Przyjmować doustnie 1 raz dziennie, niezależnie od posiłków. Klopidogrel można łączyć z kwasem acetylosalicylowym, ponieważ leki te hamują różne mechanizmy agregacji płytek krwi i w ten sposób wzmacniają wzajemne działanie (jednak przy takim połączeniu istnieje większe ryzyko powikłań krwotocznych).
W porównaniu z tiklopidyną działania niepożądane klopidogrelu są mniej wyraźne (biegunka - 4,5%, wysypka - 6%). Stosowanie klopidogrelu wiąże się z mniejszym ryzykiem wystąpienia tak poważnego powikłania jak neutropenia (0,1%), a małopłytkowość występuje rzadziej. Jako rzadkie powikłanie, podobnie jak w przypadku tiklopidyny, może rozwinąć się zakrzepowa plamica małopłytkowa.
Inhibitory fosfodiesterazy płytkowej
Dipirydamol (kurantyl*, persantyna*) został po raz pierwszy zaproponowany jako środek rozszerzający naczynia wieńcowe. Później odkryto jego zdolność do hamowania agregacji płytek krwi. Obecnie dipirydamol stosowany jest głównie jako lek przeciwpłytkowy w profilaktyce zakrzepicy. Działanie przeciwpłytkowe dipirydamolu wiąże się ze wzrostem poziomu cAMP w płytkach krwi, w wyniku czego zmniejsza się w nich stężenie cytoplazmatycznego Ca 2+. Dzieje się tak z kilku powodów. Po pierwsze, dipirydamol hamuje fosfodiesterazę, która inaktywuje cAMP. Ponadto dipirydamol hamuje wychwyt adenozyny przez komórki śródbłonka i erytrocyty oraz jej metabolizm (hamuje deaminazę adenozyny), zwiększając w ten sposób poziom adenozyny we krwi (ryc. 27-4). Adenozyna pobudza receptory płytkowe A 2 i zwiększa aktywność cyklazy adenylanowej związanej z tymi receptorami, w wyniku czego wzrasta tworzenie cAMP w płytkach krwi i zmniejsza się poziom cytoplazmatycznego Ca 2+. Dipirydamol zwiększa również poziom cAMP w komórkach mięśni gładkich naczyń, powodując rozkurcz naczyń.

Dipirydamol stosuje się w zapobieganiu udarowi niedokrwiennemu, a także chorobom tętnic obwodowych (głównie w połączeniu z kwasem acetylosalicylowym, ponieważ sam dipirydamol ma słabe działanie przeciwpłytkowe). Przepisywany doustnie 3-4 razy dziennie na 1 godzinę przed posiłkiem. W połączeniu z doustnymi lekami przeciwzakrzepowymi dipirydamol jest przepisywany w celu zapobiegania tworzeniu się zakrzepów krwi w chorobie mitralnej serca.
Podczas stosowania dipirydamolu mogą wystąpić bóle głowy, zawroty głowy, niedociśnienie tętnicze, objawy dyspeptyczne,
wysypki skórne. Ryzyko krwawienia jest mniejsze niż w przypadku stosowania kwasu acetylosalicylowego. Dipirydamol jest przeciwwskazany w przypadku dusznicy bolesnej (możliwy „zespół podkradania”).

Ryż. 27-4. Mechanizm przeciwpłytkowego działania dipirydamolu: EC – komórka śródbłonka; Receptor A2-P-adenozyny A2; PDE – fosfodiesteraza cAMP; AC – cyklaza adenylanowa; GP IIb/IIIa - glikoproteiny IIb/IIIa
Pentoksyfilina (agapuryna*, trental*), podobnie jak dipirydamol, hamuje fosfodiesterazę i zwiększa poziom cAMP. W rezultacie zmniejsza się poziom cytoplazmatycznego Ca 2 + w płytkach krwi, co prowadzi do zmniejszenia ich agregacji. Pentoksyfilina ma także inne właściwości: zwiększa odkształcalność czerwonych krwinek, zmniejsza lepkość krwi, działa rozszerzająco na naczynia, poprawiając mikrokrążenie.
Pentoksyfilinę stosuje się w leczeniu udarów mózgowo-naczyniowych, zaburzeń krążenia obwodowego różnego pochodzenia i patologii naczyniowych oczu (patrz rozdział „Leki stosowane w udarach mózgowo-naczyniowych”). Możliwe skutki uboczne: objawy dyspeptyczne, zawroty głowy, zaczerwienienie twarzy, a także obniżenie ciśnienia krwi, tachykardia, reakcje alergiczne, krwawienie. Podobnie jak dipirydamol, może wywoływać ataki podczas dławicy piersiowej.
Środki blokujące glikoproteiny IIb/IIIa błon płytek krwi
Ta grupa leków przeciwpłytkowych, które bezpośrednio oddziałują z glikoproteinami IIb/IIIa błon płytek krwi i zakłócają ich wiązanie z fibrynogenem, pojawiła się stosunkowo niedawno.
Abciximab (reopro*) - pierwszym lekiem z tej grupy jest „chimeryczne” mysie/ludzkie przeciwciało monoklonalne (fragment Fab mysich przeciwciał przeciwko glikoproteinom IIb/IIIa połączony z fragmentem Fc ludzkiej Ig). Abciximab niekonkurencyjnie hamuje wiązanie fibrynogenu z glikoproteinami IIb/IIIa na błonie płytek krwi, zaburzając ich agregację (patrz ryc. 27-3). Agregacja płytek krwi normalizuje się po 48 godzinach od podania pojedynczej dawki. Lek podaje się dożylnie (w postaci wlewu) w celu zapobiegania zakrzepicy podczas angioplastyki tętnic wieńcowych. Podczas stosowania abciximabu możliwe są krwawienia, w tym wewnętrzne (z przewodu pokarmowego, wewnątrzczaszkowe, krwawienia z dróg moczowo-płciowych), nudności, wymioty, niedociśnienie, bradykardia, reakcje alergiczne aż do wstrząsu anafilaktycznego, małopłytkowość.
Poszukiwania leków mniej uczulających, o takim samym mechanizmie działania, doprowadziły do stworzenia syntetycznych blokerów glikoprotein IIb/IIIa. Na bazie barboryny (peptydu wyizolowanego z jadu grzechotnika karłowatego) otrzymano lek ept i fi b at id (integrylina*) – cykliczny hektapeptyd imitujący sekwencję aminokwasów łańcucha fibrynogenu, który bezpośrednio wiąże się z glikoproteinami IIb/IIIa. Eptyfibatyd kompetycyjnie wypiera fibrynogen z receptorów, powodując odwracalne zaburzenie agregacji płytek krwi. Lek podaje się dożylnie w postaci wlewu; działanie przeciwpłytkowe pojawia się w ciągu 5 minut i ustępuje 6-12 godzin po zaprzestaniu podawania. Lek jest zalecany w profilaktyce tworzenia się skrzeplin podczas przezskórnej angioplastyki wieńcowej, w niestabilnej dławicy piersiowej oraz w profilaktyce zawału mięśnia sercowego. Niebezpiecznym powikłaniem podczas stosowania eptyfibatydu jest krwawienie; możliwa jest małopłytkowość.
Tirofiban (agrastat*) jest niepeptydowym blokerem glikoprotein IIb/IIIa, analogu tyrozyny. Podobnie jak eptyfibatyd, tirofiban kompetycyjnie blokuje receptory glikoproteinowe IIb/IIIa. Lek podaje się dożylnie (wlew). Szybkość początku działania, czas działania i wskazania do stosowania są takie same jak w przypadku eptyfibatydu. Skutki uboczne - krwawienie, małopłytkowość.
Aby poszerzyć możliwości stosowania leków z tej grupy, stworzono blokery glikoprotein IIb/IIIa, skuteczne przy podawaniu doustnym – xemilofiban*, sibrafiban* itp. Badania tych leków wykazały jednak ich niewystarczającą skuteczność i efekt uboczny w postać ciężkiej trombocytopenii.
27.2. LEKI WPŁYWAJĄCE NA krzepnięcie krwi
Uszkodzenie ściany naczyń inicjuje nie tylko agregację płytek krwi, ale także krzepnięcie krwi. W proces ten zaangażowanych jest wiele znanych czynników (osocze, tkanki, płytki krwi). Większość z nich to białka osocza, które krążą w stanie nieaktywnym, ale następnie są aktywowane podczas procesu krzepnięcia krwi. Aby wyjaśnić działanie leków, należy wymienić czynnik VII (prokonwertyna), czynnik IX (Boże Narodzenie), czynnik X (Stuart-Prower), czynnik II (protrombina). Te czynniki krzepnięcia są proenzymami i po aktywacji przekształcają się w enzymy proteolityczne (czynniki GCa, Xa i Na). Czynniki VIII i V po aktywacji pełnią funkcję białek kofaktorowych dla enzymów (odpowiednio czynniki IXa i Xa), zwiększając ich aktywność proteolityczną.
Czynnik VII początkowo ma niską aktywność proteolityczną, jednak w wyniku interakcji z czynnikiem tkankowym (patrz s. 481) szybko wzrasta. Aktywowany czynnik VII (czynnik VIIa) wraz z czynnikiem tkankowym i Ca 2+ tworzy kompleks, który powoduje częściową proteolizę czynników IX i X. Czynnik IXa z kolei dodatkowo aktywuje czynnik X (powstaje czynnik Xa). Czynnik Xa działa na protrombinę (czynnik II) i przekształca ją w trombinę (czynnik Ha). Trombina powoduje częściową proteolizę fibrynogenu, tworząc fibrynę (ryc. 27-5).
Proteolityczna aktywacja czynników krzepnięcia krwi ulega znacznemu przyspieszeniu, jeśli wiążą się one poprzez jony Ca 2+ z ujemnie naładowanymi fosfolipidami błon komórkowych. Fosfolipidy te pełnią rolę swego rodzaju matrycy, na której czynniki krzepnięcia łączą się w kompleksy z udziałem jonów Ca 2 +. Jednocześnie szybkość aktywacji czynników w tych kompleksach wzrasta 10 tysięcy razy lub więcej. Warunkiem koniecznym powstania takich kompleksów jest zdolność czynników II, VII, IX i X do wiązania Ca 2 +. Czynniki te zawierają ładunek ujemny
reszty kwasów γ-karboksyglutaminowych, które zapewniają ich wiązanie z Ca 2+. Tworzenie kwasów γ-karboksyglutaminowych następuje w wątrobie przy udziale witaminy K. Przy niedoborze witaminy K we krwi pojawiają się wadliwe czynniki krzepnięcia krwi II, VII, IX i X, co zakłóca tworzenie fibryny.

Ryż. 27-5. Schemat aktywacji krzepnięcia krwi w przypadku uszkodzenia ściany naczynia (Z: Katzung B. G. Farmakologia podstawowa i kliniczna. - NY, 2001, z późniejszymi zmianami): kompleksy czynników krzepnięcia krwi związane z ujemnie naładowanymi fosfolipidami błon komórkowych podkreślono literą odważna linia. Kompleks VIIa + TF + Ca 2+ aktywuje czynniki X i IX (TF - czynnik tkankowy). Kompleks IXa + VIIIa + Ca 2+ dodatkowo aktywuje czynnik X. Kompleks Xa + Va + Ca 2+ (protrombinaza) promuje konwersję protrombiny do trombiny. Czynniki zawarte w ramkach są hamowane przez heparynę
Do białek osocza krwi, które zawierają reszty kwasu γ-karboksyglutaminowego i powstają w wątrobie przy udziale witaminy K, zalicza się także białka C i S. Po aktywacji białko C (Ca) powoduje proteolityczne rozszczepienie czynników VIIIa i Va. Prowadzi to do zakłócenia tworzenia trombiny. Białko S pełni rolę kofaktora w reakcjach proteolizy. Aktywacja białka C następuje pod wpływem trombiny na powierzchni nienaruszonych (nieuszkodzonych) komórek śródbłonka, w których zachodzi ekspresja białka trombomoduliny, które jednocześnie wiąże białko C i trombinę.
27.2.1. Antykoagulanty (antykoagulanty)
Stosowane w praktyce klinicznej leki przeciwzakrzepowe albo hamują aktywne czynniki krzepnięcia bezpośrednio we krwi, albo zakłócają ich powstawanie w wątrobie. Dlatego dzieli się je na 2 grupy:
(działają bezpośrednio we krwi).
- Standard heparyny(niefrakcjonowany).
- Heparyny drobnocząsteczkowe:
enoksaparyna sodowa;
Nadroparyna wapniowa;
dalteparyna sodowa;
Rewiparyna sodowa.
- Heparynoidy:
sulodeksyd;
Danaparoid**.
- Lek antytrombina III.
- Preparaty hirudyny:
Lepirudyna*.
- Aktywowane białko C:
Drotrekogina alfa.
(hamują syntezę czynników krzepnięcia w wątrobie).
- Pochodne kumaryny:
Acenokumarol (syncumar*);
Warfaryna (warfarex*).
- Pochodne indanodionu:
Fenindion (fenylina*).
Bezpośrednie antykoagulanty
Heparyna- siarczanowany glikozaminoglikan (mukopolisacharyd), składający się z reszt D-glukozaminy i kwasu D-glukuronowego. Heparyna jest wytwarzana przez komórki tuczne w wielu tkankach; jest zawarty w dużych ilościach w wątrobie, płucach i błonie śluzowej jelit. Do celów medycznych heparynę izoluje się z błony śluzowej jelit świń i płuc bydła. W trakcie
w wyniku izolacji powstaje mieszanina frakcji o różnej długości łańcuchów polisacharydowych i różnych masach cząsteczkowych (od 3000 do 40 000 D). Frakcje o różnych masach cząsteczkowych różnią się nieznacznie aktywnością biologiczną i właściwościami farmakokinetycznymi. Dlatego też preparaty heparyny otrzymywane różnymi metodami i z różnych źródeł mogą wykazywać odmienne działanie przeciwzakrzepowe, w związku z czym konieczne jest przeprowadzenie ich standaryzacji biologicznej. O działaniu heparyny decyduje jej zdolność do wydłużania czasu krzepnięcia krwi (1 mg heparyny standardowej zawiera 130 jednostek).
Heparyna działa na czynniki krzepnięcia krwi dopiero po utworzeniu kompleksu z endogenną antykoagulantem antytrombiną III. Antytrombina III, glikoproteina osocza krwi, hamuje proteazy serynowe, do których należą czynniki krzepnięcia krwi IIa (trombina), EXa i Xa (a także XIa i XIIa). Proces inaktywacji czynnika pod wpływem samej antytrombiny III przebiega bardzo powoli. Heparyna powoduje zmiany konformacyjne w cząsteczce antytrombiny III, co prowadzi do około 1000-krotnego przyspieszenia tego procesu.

Główne działanie kompleksu heparyna-antytrombina III jest skierowane przeciwko trombinie i czynnikowi Xa, jednak mechanizmy hamowania tych czynników są pewne. Aby inaktywować trombinę, heparyna musi związać się zarówno z cząsteczką antytrombiny III, jak i cząsteczką trombiny. Jednocześnie szybka inaktywacja czynnika Xa przez kompleks heparyna-antytrombina III nie wymaga wiązania tego czynnika z heparyną. Frakcje heparyny o stosunkowo krótkim łańcuchu polimerowym (poniżej 18 jednostek sacharydowych) nie mogą jednocześnie wiązać antytrombiny III i trombiny, w związku z czym nie wykazują aktywności antytrombiny. Ich działanie wiąże się głównie z inaktywacją czynnika Xa, a co za tym idzie z zaburzeniem konwersji protrombiny do trombiny.
Oprócz wpływu na krzepnięcie krwi, heparyna ma również inne działanie: zmniejsza poziom lipidów we krwi w wyniku aktywacji lipazy lipoproteinowej (enzym ten hydrolizuje trójglicerydy) i hamuje proliferację komórek mięśni gładkich.
Heparyna po podaniu doustnym jest słabo wchłaniana, dlatego podaje się ją dożylnie, czasem podskórnie. Po podaniu dożylnym działanie następuje natychmiast i utrzymuje się przez 2-6 godzin. Po podaniu podskórnym heparyna zaczyna działać po 1-2 godzinach, czas działania wynosi 8-12 godzin (przepisywany 2-3 razy dziennie). Heparyna we krwi wiąże się z wieloma białkami, w tym z tymi, które ją neutralizują (czynnik płytkowy 4 i niektóre inne). Wysoki poziom tych białek we krwi może powodować względną oporność na lek. Ponadto heparyna wiąże się z makrofagami i komórkami śródbłonka, powodując jej degradację (depolimeryzację). Heparyna jest również metabolizowana w wątrobie i wydalana przez nerki.
Heparynę stosuje się w profilaktyce i leczeniu zakrzepicy żył głębokich i zatorowości płucnej, niestabilnej dławicy piersiowej i zawale mięśnia sercowego, zapobieganiu zakrzepicy tętnic obwodowych, wymianie zastawek serca i krążeniu pozaustrojowemu. Heparynę podaje się w jednostkach działania (AU).
Najczęstszym powikłaniem leczenia heparyną są krwawienia, które mogą być spowodowane zahamowaniem czynności płytek krwi lub zmniejszeniem ich liczby (trombocytopenia). Wiązanie heparyny z czynnikiem von Willebranda wydaje się wyjaśniać jej hamujący wpływ na adhezję i agregację płytek krwi. W takich przypadkach odstawia się heparynę, a w przypadku poważnego krwawienia podaje się także dożylnie siarczan protaminy, który neutralizuje heparynę tworząc nierozpuszczalny kompleks.
Małopłytkowość występuje w 7-14 dniu leczenia u około 1-5% pacjentów otrzymujących heparynę. Jest to spowodowane pojawieniem się przeciwciał (IgG) skierowanych przeciwko kompleksowi heparyna-czynnik płytkowy 4. Kompleks ten wiąże się z błoną płytek krwi po neutralizacji heparyny przez czynnik 4, glikoproteinę pochodzącą z płytek krwi, która jest uwalniana podczas agregacji płytek krwi. U mniej niż 1% pacjentów z małopłytkowością występuje zakrzepica spowodowana uszkodzeniem śródbłonka
komórek i aktywację płytek krwi przez przeciwciała przeciwko kompleksowi heparyna-czynnik 4. Stan ten wymaga odstawienia heparyny i zastosowania leków przeciwzakrzepowych nie powodujących małopłytkowości: danaparoidu** i lepirudyny**.
Przy długotrwałym stosowaniu heparyny (ponad 3 miesiące) może rozwinąć się osteoporoza. Należy to szczególnie wziąć pod uwagę przepisując heparynę kobietom w ciąży. Hiperkaliemia związana z hamowaniem syntezy aldosteronu w nadnerczach jest dość rzadkim powikłaniem leczenia heparyną.
Heparyny drobnocząsteczkowe (frakcjonowane). składają się z fragmentów heparyny o masie cząsteczkowej od 1000 do 10 000 D (średnio 4000-5000 D). Otrzymuje się je poprzez frakcjonowanie, hydrolizę lub depolimeryzację konwencjonalnej (niefrakcjonowanej) heparyny. Leki te, podobnie jak heparyna, działają na czynniki krzepnięcia poprzez antytrombinę III, ale różnią się od heparyny następującymi właściwościami:
Hamują aktywność czynnika Xa w większym stopniu niż czynnika IIa (3-4 razy);
Mają większą biodostępność po podaniu podskórnym (heparyny drobnocząsteczkowe – ok. 90%, heparyna standardowa – 20%);
Działają dłużej, co pozwala na ich podawanie 1-2 razy dziennie;
Mają mniejsze powinowactwo do czynnika płytkowego 4, więc rzadziej powodują trombocytopenię niż standardowa heparyna;
Rzadziej powodują osteoporozę.
W praktyce krajowej stosuje się następujące preparaty heparyny drobnocząsteczkowej: en o x a paryna sodowa (Clexane*), nadroparyna wapniowa (Fraxiparin*), d alte paryna sodowa (Fragmin*), reviparin sodowy (klivarin*). Leki te są niejednorodne w swoim składzie (zawierają różne frakcje heparyny), dlatego różnią się nieco od siebie właściwościami fizykochemicznymi, farmakokinetycznymi i działaniem.
Heparyny drobnocząsteczkowe stosowane są w profilaktyce i leczeniu zakrzepicy żył głębokich (zwłaszcza po operacjach), w profilaktyce zatorowości płucnej, a także w niestabilnej dławicy piersiowej i zawale mięśnia sercowego. W profilaktyce wskazane są preparaty heparyny drobnocząsteczkowej
tiki i leczenie zakrzepicy w praktyce położniczej. Podawać wyłącznie podskórnie. Dawkowanie podawane jest w ME (jednostkach międzynarodowych).
Heparyny drobnocząsteczkowe, podobnie jak preparaty heparyny niefrakcjonowanej, mogą powodować krwawienie. W pierwszych dniach leczenia możliwa jest umiarkowana małopłytkowość. Heparyny drobnocząsteczkowe w niektórych przypadkach zwiększają aktywność enzymów wątrobowych i mogą powodować reakcje alergiczne. Siarczan protaminy nie eliminuje całkowicie działania heparyny drobnocząsteczkowej.
Ostatnio w praktyce klinicznej pojawił się lek fond a - rinux sodu - syntetyczny pentasacharyd, który wiążąc się z antytrombiną III, przyspiesza inaktywację czynnika Xa. Lek produkowany w postaci soli sodowej i stosowany w profilaktyce zakrzepicy żylnej i zatorowości płucnej w chirurgii ortopedycznej.
Heparynoidy- siarczanowane glikozaminoglikany, strukturalnie podobne do heparyn. Podobnie jak heparyna, wzmacniają hamujące działanie antytrombiny III na czynniki krzepnięcia krwi. Pod wieloma istotnymi cechami różnią się od heparyny i heparyny drobnocząsteczkowej, dlatego zalicza się je do specjalnej grupy. Do tej grupy należą Danaparoid* i sulodeksyd. Leki te otrzymywane są z błony śluzowej jelit świń.
Danaparoid** (organon**) zawiera mieszaninę siarczanu heparanu, siarczanu dermatanu i siarczanu chondroityny. Danaparoid p hamuje czynnik Xa w większym stopniu niż protrombina. Lek podaje się podskórnie w celu zapobiegania i leczenia zakrzepicy żylnej. Danaparoid p nie wiąże się z czynnikiem płytkowym 4 i nie powoduje małopłytkowości. Dlatego jest wskazany w przypadkach, gdy leczenie heparyną jest powikłane małopłytkowością.
Sulodeksyd (Wessel Due F*) składa się z mieszaniny siarczanu heparanu i siarczanu dermatanu. Sulodeksyd w dużym stopniu zmniejsza aktywność czynnika Xa, z niewielkim wpływem na protrombinę. Lek zwiększa aktywność fibrynolityczną, działa ochronnie na śródbłonek naczyń, ma właściwości hipolipidemiczne. Sulodeksyd jest wskazany w leczeniu chorób naczyń obwodowych ze zwiększonym ryzykiem zakrzepicy. Istnieją postacie dawkowania leku do podawania pozajelitowego (dożylnego i domięśniowego) oraz do podawania doustnego.
Antytrombina IIIniezbędny do przeciwzakrzepowego działania heparyny, heparyny drobnocząsteczkowej i heparynoidów.
W przypadku dziedzicznego niedoboru antytrombiny III jej lek stosuje się i podaje dożylnie. Przy długotrwałym stosowaniu heparyny zwiększa się spożycie antytrombiny III, przez co jej stężenie we krwi wyraźnie maleje. Zmniejsza to skuteczność terapii heparyną. W takich przypadkach podaje się także antytrombinę III.
Hirudin- białko o masie cząsteczkowej 7 kDa, odkryte po raz pierwszy w gruczołach ślinowych pijawek lekarskich Hirudo leki. Hirudyna, podobnie jak heparyna, jest antykoagulantem działającym bezpośrednio we krwi, jednak w przeciwieństwie do heparyny, hirudyna bezpośrednio hamuje trombinę: selektywnie wiąże się z nią i inaktywuje ją bez udziału antytrombiny III. Zahamowanie jest nieodwracalne. W przeciwieństwie do heparyny, hirudyna ma zdolność hamowania trombiny związanej z skrzepami krwi, a tym samym opóźniania wzrostu skrzepów krwi. Hirudyna nie wchodzi w interakcję z czynnikiem płytkowym 4 i dlatego nie powoduje małopłytkowości.
Do zastosowania klinicznego otrzymano rekombinowany preparat hirudyny - lepirudin* (refludan*). Zaleca się stosować w celu zapobiegania możliwym powikłaniom zakrzepowo-zatorowym w małopłytkowości wywołanej heparyną. Lepirudynę* podaje się dożylnie. Podczas stosowania może wystąpić krwawienie. Nie ma swoistego antidotum na preparaty hirudyny.
Drotrekogina alfa (Zigris*) jest rekombinowanym preparatem aktywowanego białka C. Hamuje tworzenie trombiny, powodując proteolityczną inaktywację czynników krzepnięcia krwi VIIIa i Va. Ponadto drotrekogina zwiększa aktywność fibrynolityczną osocza krwi, zmniejszając ilość krążącego we krwi inhibitora aktywatora plazminogenu typu 1. Obecność działania przeciwzapalnego leku jest związana z jego hamującym wpływem na uwalnianie czynnika martwicy nowotworu alfa z monocytów. Wszystkie te właściwości drotrekoginy decydują o jej skuteczności w leczeniu wstrząsu septycznego (głównymi objawami tego schorzenia są stany zapalne i zwiększona krzepliwość krwi). Podobnie jak inne leki przeciwzakrzepowe, lek może powodować powikłania krwotoczne.
Pośrednie antykoagulanty
Leki te w odróżnieniu od heparyny nie wpływają bezpośrednio na czynniki krzepnięcia krwi. Hamują syn-
synteza w wątrobie białek osocza krwi zależna od witaminy K – czynnik II (protrombina), czynniki VII, IX i X (patrz ryc. 27-5). Witamina K jest niezbędna do tworzenia funkcjonalnie cennych czynników, gdyż pełni funkcję koenzymu w reakcji γ-karboksylacji reszt kwasu glutaminowego. Zredukowana forma witaminy K, hydrochinon, wykazuje działanie koenzymowe. Podczas karboksylacji hydrochinon witaminy K ulega utlenieniu, tworząc nieaktywny epoksyd witaminy K. Pośrednie antykoagulanty zapobiegają konwersji (redukcji) nieaktywnego epoksydu witaminy K do aktywnej witaminy K-hydrochinonu przez reduktazę epoksydową i DT-diaforazę, hamując te enzymy. Dlatego zalicza się je do antagonistów witaminy K (ryc. 27-6).

Ryż. 27-6.Mechanizm działania witaminy K i pośrednich antykoagulantów
Pośrednie antykoagulanty nie powodują natychmiastowego zmniejszenia stężenia czynników krzepnięcia we krwi. Ich działanie charakteryzuje się okresem utajonym. Zatem działanie przeciwzakrzepowe acenokumarolu osiąga maksymalną wartość po 48 godzinach lub dłużej. Ten powolny rozwój efektu można wytłumaczyć faktem, że po podaniu tych leków pełnowartościowe czynniki krzepnięcia krążą jeszcze przez pewien czas we krwi (szybkość wystąpienia efektu zależy od czasu, w którym czynniki krzepnięcia kompleks protrombiny ulegają degradacji). Działanie pośrednich antykoagulantów trwa około 2-4 dni, leki mają zdolność kumulacji.

Pośrednie leki przeciwzakrzepowe stosowane są w długotrwałej profilaktyce i leczeniu zakrzepicy i choroby zakrzepowo-zatorowej (zakrzepica żył głębokich, zatorowość płucna, powikłania zakrzepowo-zatorowe w migotaniu przedsionków, zawale mięśnia sercowego, wymiana zastawki serca), w praktyce chirurgicznej w celu zapobiegania tworzeniu się skrzeplin w okresie pooperacyjnym. Wprowadzono do środka. Leczenie odbywa się pod obowiązkowym monitorowaniem poziomu protrombiny w osoczu krwi poprzez oznaczenie czasu protrombinowego – wskaźnika, którego wartość zależy od zawartości protrombiny we krwi oraz czynników IX i X.
Najczęstszym powikłaniem podczas stosowania pośrednich antykoagulantów jest krwawienie. Ryzyko krwawienia wzrasta w przypadku jednoczesnego stosowania aspiryny* i innych leków przeciwpłytkowych. W celu zatamowania krwawień wywołanych pośrednimi antykoagulantami należy podawać preparaty witaminy K1 oraz koncentrat kompleksu protrombiny (zawiera czynniki II, VII, IX i X). Możliwe są inne działania niepożądane: reakcje alergiczne, biegunka, zaburzenia czynności wątroby, martwica skóry. Pośrednie leki przeciwzakrzepowe są przeciwwskazane w czasie ciąży: przenikają przez łożysko i mogą działać teratogennie (zaburzają tworzenie kośćca, gdyż
hamują tworzenie osteokalcyny, białka kości zależnego od witaminy K). Fenindion (fenilina*) może powodować hamowanie hematopoezy.
27.2.2. Środki krzepnięcia krwi
Leki zwiększające krzepliwość krwi stosuje się w celu zatrzymania krwawienia, dlatego zalicza się je do środków hemostatycznych (hemostatycznych) lub przeciwkrwotocznych. Do tej grupy zaliczają się substancje niezbędne do tworzenia czynników krzepnięcia krwi (preparaty witaminy K) oraz preparaty samych czynników krzepnięcia.
Preparaty witaminy K
Witamina K występuje w dwóch postaciach – witaminy K 1 (filochinon), występującej w roślinach oraz witaminy K 2 – grupy związków (menachinonów) syntetyzowanych przez mikroorganizmy (w szczególności mikroflorę jelitową człowieka). Witaminy K 1 i K 2 to związki rozpuszczalne w tłuszczach, pochodne 2-metylo-1,4-naftochinonu, różniące się długością i charakterem bocznego łańcucha węglowego. Witamina Kj pozyskiwana jest syntetycznie, jej preparat nazywany jest fitomenadionem. Zsyntetyzowano rozpuszczalny w wodzie prekursor witaminy K, 2-metylo-1,4-naftochinon (menadion), który wykazuje działanie prowitaminowe. Związek ten nazwano witaminą K3. Pochodna witaminy K3 – wodorosiarczyn sodu menadionu – stosowana jest w praktyce lekarskiej pod nazwą b i k a - sol*.
Witamina K jest niezbędna do syntezy w wątrobie protrombiny (czynnik II) oraz czynników krzepnięcia krwi VII, IX i X, a także białek C i S. Wiadomo, że witamina K uczestniczy w syntezie białka tkanki kostnej – osteokalcyny.
Struktura wszystkich białek zależnych od witaminy K ma wspólną cechę: białka te zawierają reszty kwasu γ-karboksyglutaminowego, które wiążą jony Ca 2+. Witamina K-hydrochinon pełni funkcję koenzymu w reakcji γ-karboksylacji reszt kwasu glutaminowego (patrz ryc. 27-6). Przy niedoborze witaminy K pojawiają się nieaktywne prekursory czynników krzepnięcia krwi, które nie są w stanie wiązać Ca 2+. Niedobór witaminy K w organizmie najszybciej prowadzi do upośledzenia hemokoagulacji. Dlatego główny
a najwcześniejszymi objawami niedoboru witaminy K są krwawienie i krwotok.
Preparaty witaminy K stosuje się w celu zapobiegania i tamowania krwawień i innych powikłań krwotocznych spowodowanych niedoborem witaminy K w organizmie, np. zespołem krwotocznym noworodków. Niedobór witaminy K u noworodków może być spowodowany zarówno niewystarczającym spożyciem witaminy K 1, jak i brakiem mikroflory jelitowej syntetyzującej witaminę K 2 . Aby zapobiec takim powikłaniom, zaleca się profilaktyczne podawanie witaminy K 1 noworodkom w pierwszych godzinach życia.
Preparaty witaminy K są wskazane w przypadku zmniejszonego wchłaniania witaminy K w jelitach na skutek upośledzonego wydzielania żółci w żółtaczce zaporowej (żółć jest niezbędna do wchłaniania witaminy K rozpuszczalnej w tłuszczach) lub przy zespole złego wchłaniania (wlew, zapalenie jelit, choroba Leśniowskiego-Crohna itp.) .)
Preparaty witaminy K 1 są skuteczne w przypadku krwawień wywołanych pośrednimi antykoagulantami. Podaje się je powoli doustnie i dożylnie.
Preparaty witaminy K mogą powodować reakcje alergiczne (wysypka, swędzenie, rumień, skurcz oskrzeli). Po podaniu dożylnym istnieje ryzyko reakcji rzekomoanafilaktycznych. Stosowanie preparatów witaminy K3 (Vikasol*) u noworodków wiąże się z ryzykiem rozwoju niedokrwistości hemolitycznej i hiperbilirubinemii.
Preparaty czynników krzepnięcia
Zapotrzebowanie na takie leki pojawia się, gdy brakuje jednego lub więcej czynników krzepnięcia krwi.
Antyhemofilowy czynnik krzepnięcia krwi VIII (hemophil M*, immunat* itp.) jest suchym koncentratem czynnika VIII. Leki otrzymywane są z osocza krwi dawcy poddanego podwójnej inaktywacji wirusa i standaryzowane na zawartość czynnika VIII. Są bardziej aktywne i bezpieczniejsze niż krioprecypitat*.
Krioprecypitat* to koncentrat białek osocza krwi, w skład którego wchodzi czynnik VIII, czynnik von Willebranda, fibronektyna, a także w mniejszym stopniu inne czynniki krzepnięcia krwi i niewielkie ilości fibrynogenu.
Leki podaje się dożylnie w przypadku dziedzicznego (hemofilia A) i nabytego niedoboru czynnika VIII. krioprecypitat*,
ponadto stosowany jest w terapii zastępczej choroby von Willebranda (dziedziczny niedobór czynnika von Willebranda) i afibrynogenemii. Po podaniu możliwe są działania niepożądane w postaci tachykardii, niedociśnienia tętniczego, duszności, reakcji alergicznych (pokrzywka, gorączka, wstrząs anafilaktyczny), a także hemolizy czerwonych krwinek.
Wszystkie preparaty czynników krzepnięcia otrzymywane z osocza krwi mają istotną wadę - możliwość przenoszenia infekcji wirusowych (HIV, zapalenie wątroby). Obecnie uzyskano rekombinowane preparaty czynnika VIII i czynnika von Willebranda, których zastosowanie zmniejsza ryzyko infekcji.
Oprócz leków na czynniki krzepnięcia, w łagodnych postaciach hemofilii A i choroby von Willebranda stosuje się desmopresynę, analog wazopresyny argininowej. Desmopresyna zwiększa zawartość czynnika von Willebranda w osoczu krwi, ułatwiając jego uwalnianie z komórek śródbłonka i zwiększa aktywność czynnika
VIII. Lek podaje się pozajelitowo.
Czynnik krzepnięcia krwi IX (agemphil B*, immunonina*, oktanina*) jest oczyszczoną frakcją ludzkiego osocza wzbogaconą w czynnik IX. Stosowany przy wrodzonym (hemofilia B) i nabytym niedoborze czynnika IX, a także przy przedawkowaniu pośrednich antykoagulantów. Skutki uboczne są takie same jak w przypadku leków zawierających czynnik VIII.
Aktywowany Eptakog alfa (novoseven *) jest rekombinowanym czynnikiem krzepnięcia krwi VIIa. Stosowany przy niedoborze czynnika VII i innych czynników krzepnięcia (V, II,
IX, X).
Miejscowo, aby zatrzymać krwawienie z małych naczyń włosowatych i narządów miąższowych, stosuje się trombinę (uzyskaną z osocza krwi dawcy), a także gąbki hemostatyczne (kolagen, żelatyna).
Aby zatamować krwawienia z macicy, płuc, nerek, jelit i innych, stosuje się lecznicze preparaty roślinne: liście pokrzywy, ziele krwawnika, ziele rdestu pieprzowego, ziele rdestu rdestowego, kora kaliny, kwiaty arniki, odurzający lagochilus. Rośliny lecznicze stosuje się w postaci naparów, nalewek i ekstraktów wewnętrznie i miejscowo.
27.3. LEKI WPŁYWAJĄCE NA FIBRYNOLIZĘ
Kiedy tworzą się skrzepy krwi, aktywowany jest układ fibrynolityczny, który zapewnia rozpuszczenie (lizę) fibryny i zniszczenie skrzepu krwi. Prowadzi to do przywrócenia prawidłowego przepływu krwi.
W procesie fibrynolizy nieaktywny plazminogen ulega przemianie do plazminy (fibrynolizyny) przy udziale aktywatorów plazminogenu. Plazmina hydrolizuje fibrynę, tworząc rozpuszczalne peptydy. Plazmina nie ma swoistości i powoduje także zniszczenie fibrynogenu i niektórych innych czynników krzepnięcia krwi, co zwiększa ryzyko krwawień. Plazmina krążąca we krwi jest szybko inaktywowana przez α2-antyplazminę i inne inhibitory i dlatego zwykle nie ma ogólnoustrojowego działania fibrynogenolitycznego. Jednak w pewnych stanach patologicznych lub przy stosowaniu środków fibrynolitycznych możliwa jest nadmierna aktywacja plazminogenu w osoczu, co może powodować krwawienie.
27.3.1. Środki fibrynolityczne (trombolityczne).
Środki fibrynolityczne stosuje się do rozpuszczania skrzepów krwi w zakrzepicy wieńcowej (ostrym zawale mięśnia sercowego), zakrzepicy żył głębokich, ostrej zakrzepicy tętnic obwodowych i zatorowości płucnej.
Jako środki fibrynolityczne stosuje się preparaty aktywujące plazminogen: preparaty streptokinazy, preparaty tkankowych aktywatorów plazminogenu, preparaty urokinazy.
Leki streptokinazowe
Streptokinaza (kabikinaza*) jest wysoce oczyszczonym preparatem białkowym otrzymywanym z hodowli paciorkowców β-hemolizujących. Streptokinaza uzyskuje aktywność proteolityczną dopiero w połączeniu z plazminogenem. Po podaniu streptokinazy tworzy się równomolowy kompleks streptokinaza-plazminogen, przekształcający plazminogen w plazminę. Streptokinaza działa na plazminogen zarówno w skrzepie krwi, jak i w osoczu krwi (ryc. 27-7).
Streptokinazę podaje się dożylnie w ostrym zawale mięśnia sercowego spowodowanym zakrzepicą naczyń wieńcowych (większość
skuteczny przez pierwsze 3-6 godzin), w zakrzepicy żył głębokich, zatorowości płucnej i zakrzepicy naczyń siatkówki. Streptokinazę podaje się w ME (jednostkach międzynarodowych).
Częstymi powikłaniami podczas stosowania streptokinazy są krwawienia, które można wiązać zarówno z aktywacją krążącego we krwi plazminogenu (powstająca plazmina niszczy fibrynogen, co skutkuje zmniejszoną agregacją płytek krwi), jak i z rozpuszczeniem fizjologicznych skrzepów krwi. Możliwe są nudności, wymioty i niedociśnienie tętnicze. Ze względu na obecność właściwości antygenowych streptokinaza może powodować reakcje alergiczne, w tym wstrząs anafilaktyczny. Ich niebezpieczeństwo wzrasta wraz z wielokrotnym podawaniem leku. Przeciwciała krążące we krwi mogą inaktywować streptokinazę i zmniejszać skuteczność terapii.
Anistreplaza** (eminaza**) jest kompleksem streptokinazy z plazminogenem acylowanej lizyny. Grupa acylowa w cząsteczce plazminogenu zamyka miejsce katalityczne, co zapobiega aktywacji plazminogenu. Lek jest prolekiem i nabywa zdolność przekształcania plazminogenu w plazminę dopiero po rozszczepieniu grupy acylowej. Szybkość deacylowania, a tym samym czas tworzenia aktywnego leku, zależy od charakteru grupy acylowej i może wynosić od 40 minut do kilku godzin. Anistreplazę** podaje się dożylnie. Po jednorazowym wstrzyknięciu efekt fibrynolityczny utrzymuje się przez 4-6 godzin. Wskazania do stosowania i skutki uboczne są takie same jak w przypadku streptokinazy.
Tkankowy aktywator plazminogenu i preparaty urokinazy
Głównymi fizjologicznymi aktywatorami plazminogenu są tkankowy aktywator plazminogenu i urokinaza.
Tkankowy aktywator plazminogenu jest wytwarzany przez komórki śródbłonka. Powoduje częściową proteolizę plazminogenu, w wyniku czego następuje jego konwersja do plazminy. Charakterystyczną cechą aktywatora tkankowego jest jego wysokie powinowactwo do fibryny, co setki razy przyspiesza jego działanie na plazminogen. W rezultacie aktywator tkankowy aktywuje z większą szybkością cząsteczki plazminogenu, które są zaadsorbowane na włóknach fibrynowych. Zatem działanie tkankowego aktywatora plazminogenu jest ograniczone przez fibrynę skrzeplinową. Dostając się do krwioobiegu,

Ryż. 27-7.Mechanizm działania środków fibrynolitycznych: tPA – tkankowy aktywator plazminogenu; FDP – produkty degradacji fibrynogenu; EC - komórka śródbłonka; ? - aktywacja; Θ - liza
aktywator tkankowy wiąże się ze specyficznym inhibitorem, dlatego ma niewielki wpływ na krążący we krwi plazminogen i w mniejszym stopniu obniża poziom fibrynogenu.
Do użytku klinicznego otrzymano rekombinowane preparaty tkankowego aktywatora plazminogenu: alteplazę (actilyse*) i tenekteplazę (metalise*). Leki podaje się dożylnie w przypadku ostrego zawału mięśnia sercowego spowodowanego zakrzepicą naczyń wieńcowych (skuteczne w ciągu pierwszych 6-12 godzin), w przypadku zatorowości płucnej. Pomimo tego, że alteplaza ma niewielki wpływ na krążący we krwi plazminogen, przy jej stosowaniu często występują powikłania krwotoczne. Nie ma właściwości antygenowych. Tenekteplaza ma zwiększoną specyficzność wobec fibryny skrzepliny.
Urokinaza jest wytwarzana przez komórki nerek i występuje w moczu. W nerkach powstaje jednołańcuchowa urokinaza (prourokinaza), która pod wpływem plazminy przekształca się w formę aktywną – dwułańcuchową urokinazę. Urokinaza dwułańcuchowa działa bezpośrednio aktywująco na plazminogen (nie jest wymagane tworzenie kompleksu z plazminogenem). Dwułańcuchowy preparat urokinazy otrzymywany jest z hodowli ludzkich embrionalnych komórek nerek. Stosowany w ostrym zawale mięśnia sercowego, zakrzepicy żylnej i tętniczej, zatorowości płucnej. Podawany dożylnie. Dozowany we mnie. W porównaniu do tkankowego aktywatora plazminogenu, urokinaza działa w większym stopniu na plazminogen krążący we krwi, w efekcie powstająca we krwi plazmina powoduje rozkład fibrynogenu (patrz ryc. 27-7). Jednocześnie zmniejsza się agregacja płytek krwi i powstają produkty degradacji fibrynogenu, które działają przeciwzakrzepowo. Głównymi działaniami niepożądanymi są krwawienie. Nie posiada właściwości antygenowych.
Otrzymano rekombinowany preparat jednołańcuchowej urokinazy (prourokinazy) - c a p u p l a z a * - który wykazuje większą specyficzność wobec fibryny skrzepliny niż urokinaza.
27.3.2. Środki antyfibrynolityczne
Leki antyfibrynolityczne stosuje się w celu tamowania krwawień spowodowanych wzmożoną aktywnością układu fibrynolitycznego podczas urazów, zabiegów chirurgicznych, porodu,
choroby wątroby, zapalenie gruczołu krokowego, krwotok miesiączkowy, a także przedawkowanie leków fibrynolitycznych. W tym celu stosuje się leki hamujące aktywację plazminogenu lub będące inhibitorami plazminy.
Kwas aminokapronowy wiąże się z plazminogenem i zapobiega jego konwersji do plazminy. Ponadto zaburza działanie plazminy na fibrynę. Lek podaje się doustnie i dożylnie. Możliwe skutki uboczne: niedociśnienie tętnicze, bradykardia, zaburzenia rytmu, zawroty głowy, nudności, biegunka. Podobne działanie wykazuje kwas aminometylobenzoesowy (Ambene*, Pamba*).
Kwas traneksamowy (traneksam*, cyklokapron*) hamuje aktywację plazminogenu. Lek podaje się doustnie i dożylnie. Jest skuteczniejszy niż kwas aminokapronowy i działa dłużej. Działania niepożądane obejmują objawy dyspeptyczne (anoreksja, nudności, wymioty, biegunka), zawroty głowy, senność; Możliwe są reakcje alergiczne skóry.
Aprotynina (Gordox*, Contrical*, Trasylol*, Ingitril*) hamuje plazminę i inne enzymy proteolityczne. Lek podaje się dożylnie. Skutki uboczne: niedociśnienie tętnicze, tachykardia, nudności, wymioty, reakcje alergiczne.
Płytki krwi, najmniejsze krwinki (w porównaniu do leukocytów i czerwonych krwinek), pełnią najważniejszą funkcję - chronią organizm przed utratą krwi. Agregacja płytek krwi to proces sklejania się komórek, będący początkowym etapem tworzenia skrzepu krwi.
Drugi etap polega na przyczepieniu się płytek krwi do ściany uszkodzonego naczynia. Nici fibrynowe, inne elementy i nowe przylegające komórki są nakładane warstwami na masę płytek krwi. W ten sposób skrzeplina powiększa się do rozmiarów zdolnych do zablokowania średnicy naczynia i zatrzymania krwawienia. Czasami od szybkości procesu zależy życie człowieka.
Rola agregacji płytek krwi w procesie krzepnięcia krwi
Krzepnięcie krwi zależy od wielu czynników. Jeden z nich, agregacja płytek krwi, w zdrowym organizmie ma charakter ochronny i adaptacyjny. Komórki sklejają się tylko w krwawiącym naczyniu. W tym przypadku proces odgrywa pozytywną rolę.
Znane są jednak stany patologiczne, w których tworzenie się skrzepów krwi jest niepożądane, ponieważ prowadzi to do zakłócenia odżywiania ważnych narządów. Na przykład z zawałem mięśnia sercowego, udarem, zakrzepicą tętnic wiodących. Aktywność agregacyjna płytek krwi występuje po stronie zmian patologicznych. Trzeba z tym walczyć za pomocą różnych leków.
Istnieje praktyczna potrzeba ilościowego określenia dobrej i złej agregacji płytek krwi. Aby to zrobić, musisz użyć normy i rozróżnić odchylenia.
Jak określić normalność i patologię?
Czy badanie krwi może wykazać zdolność agregacji płytek krwi u konkretnej osoby? Przecież do przeprowadzenia badania pobierana jest krew z żyły i od tego momentu „rozkazy” organizmu nie oddziałują na krwinki. Ten rodzaj analizy nazywany jest „in vitro”, co jest dosłownym tłumaczeniem z łaciny „na szkle, w probówce”. Naukowcy zawsze starają się badać reakcję w warunkach zbliżonych do ludzkiego ciała. Dopiero uzyskane w ten sposób dane można uznać za wiarygodne i wykorzystać w diagnostyce.
Zdolność płytek krwi określa się na podstawie indukowanej agregacji. Oznacza to, że jako substancję indukującą stosuje się substancje, które nie są obce organizmowi pod względem chemicznym i mogą powodować powstawanie zakrzepów. Jako induktory wykorzystywane są składniki ściany naczyń: difosforan adenozyny (ADP), ristocetyna (rystomycyna), kolagen, serotonina, kwas arachidonowy, adrenalina.
Agregację spontaniczną określa się bez induktorów.
Techniki kwantyfikacji opierają się na przepuszczaniu fal świetlnych przez osocze krwi bogate w płytki krwi. Stopień aktywności agregacyjnej bada się na podstawie różnicy gęstości światła plazmy przed rozpoczęciem koagulacji i po uzyskaniu maksymalnego wyniku. Określana jest także szybkość agregacji w pierwszej minucie, charakter i kształt fal.
Szybkość zależy od substancji induktora i jej stężenia.
Agregację płytek krwi za pomocą ADP zwykle przepisuje się i ocenia w skojarzeniu z kolagenem, ristomycyną i adrenaliną.
Norma dla analizy za pomocą ADP wynosi od 30,7 do 77,7%. Stopień agregacji płytek krwi pod wpływem adrenaliny waha się od 35 do 92,5%. W badaniu z kolagenem za normalne uważa się wartości od 46,4 do 93,1%.
Zasady przygotowania do analizy
Aby wykonać badanie krwi pod kątem zdolności agregacji, musisz zrozumieć, że test będzie niedokładny, jeśli zostaną naruszone zasady przygotowania. We krwi będą substancje, które wpływają na wynik.
- Na tydzień przed oddaniem krwi należy odstawić wszystkie leki zawierające aspirynę, dipirydamol, indometacynę, sulfapirydazynę i leki przeciwdepresyjne. Stosowanie tych leków hamuje (hamuje) powstawanie skrzeplin. Jeżeli nie można przerwać stosowania leku, należy poinformować o tym technika laboratoryjnego.
- Nie powinieneś jeść przez co najmniej 12 godzin; tłuste potrawy szczególnie wpływają na Twoje wyniki.
- Pacjent powinien zachować jak największy spokój i nie wykonywać pracy fizycznej.
- Dzień wcześniej wyklucz z pożywienia kawę, alkohol, czosnek i nie pal.
- Analizy nie przeprowadza się, jeśli występuje aktywny proces zapalny.
Zbite płytki krwi są widoczne pod mikroskopem
Badanie krwi na agregację płytek krwi zleca lekarz, jeśli konieczne jest leczenie lekami przeciwzakrzepowymi, monitorując ich skuteczność, dobierając optymalną dawkę, aby zdiagnozować zwiększone krwawienie.
Dekodowanie wyników
Powody prowadzenia badań z trzema standardowymi induktorami jednocześnie i w razie potrzeby dodawania nowych, stanowią dominujący mechanizm aktywacji jednego z czynników krzepnięcia. Zidentyfikowana zmieniona norma, np. w przypadku ADP przy braku dynamiki z innymi cewkami, ma znaczenie diagnostyczne. Wyniki ocenia lekarz.
Zmniejszona agregacja płytek krwi może być spowodowana:
- skuteczne stosowanie terapii przeciwpłytkowej;
- grupa chorób zwanych trombocytopatiami.

Aparatura do analizy agregacji
Rola trombocytopatii
Trombocytopatie mogą być dziedziczne lub nabyte w wyniku innych chorób. Statystyki mówią, że na tę patologię cierpi nawet 10% populacji planety. Wszystkie są związane z dysfunkcją płytek krwi w akumulacji niektórych substancji.
W rezultacie nie dochodzi do krzepnięcia i tworzenia się skrzepów krwi, co prowadzi do zwiększonego krwawienia w przypadku małych ran i siniaków (krwawienie wewnętrzne).
Choroby pojawiają się już w dzieciństwie z krwawiącymi dziąsłami, częstymi krwawieniami z nosa, licznymi siniakami na ciele dziecka, obrzękami stawów spowodowanymi siniakami. W okresie dojrzewania dziewczęta zaczynają mieć długie i obfite miesiączki. Krwawienie prowadzi do rozwoju anemii (niedokrwistości).
Niską zdolność agregacji w trombocytopatii mogą aktywować infekcje wirusowe i bakteryjne, leki i procedury fizjoterapeutyczne.

Krwawienia z nosa w 80% przypadków są spowodowane trombocytopatią, a tylko w 20% chorobami narządów laryngologicznych
Trombocytopatie wtórne
Objawowe (wtórne) trombocytopatie powstają w przewlekłej białaczce, szpiczaku. Stan ten jest typowy dla końcowego stadium niewydolności nerek (mocznicy), upośledzenia czynności tarczycy.
Chirurdzy spotykają się z trombocytopatią, gdy podczas zabiegów chirurgicznych występuje zwiększone krwawienie.
Zwiększenie agregacji płytek krwi obserwuje się w przypadku:
- powszechna miażdżyca naczyń;
- nadciśnienie;
- zawał narządów wewnętrznych;
- zakrzepica tętnic brzusznych;
- udar mózgu;
- cukrzyca
Zmiany agregacji podczas ciąży
Agregacja płytek krwi w czasie ciąży może odbiegać od wartości prawidłowych.
Agregacja jest zmniejszona z powodu niewystarczającej produkcji płytek krwi lub naruszenia ich składu jakościowego. Objawia się to krwawieniem i siniakami. Podczas porodu należy wziąć pod uwagę możliwość wystąpienia masywnego krwawienia.
Agregacja jest zwiększona najczęściej podczas zatrucia z powodu utraty płynów w wyniku wymiotów i biegunki. Wzrost stężenia we krwi prowadzi do zwiększonego tworzenia się skrzepliny. Może to prowadzić do wczesnego poronienia. Umiarkowana hiperagregacja jest uważana za normalną w czasie ciąży i wiąże się z rozwojem krążenia łożyskowego.
- w przypadku poronienia;
- leczenie niepłodności;
- przed i w trakcie stosowania środków antykoncepcyjnych;
- przed planowaną ciążą.
Analiza właściwości agregacyjnych płytek krwi pozwala na identyfikację zagrożeń, przewidywanie groźnych powikłań w przebiegu chorób i szybkie wdrażanie terapii zapobiegawczej.
Płytki krwi to komórki krwi, które nie mają koloru. Pełnią ważną funkcję w organizmie, chroniąc go przed utratą krwi. Proces ten polega na agregacji płytek krwi we krwi, ma swoje własne standardowe wskaźniki.
Aby zrozumieć, co to jest, musisz mieć pojęcie o tworzeniu się skrzepu krwi, istniejących standardach i niebezpieczeństwie odchyleń od normalnych wartości.
Opis i rola w organizmie człowieka
Po uszkodzeniu tkanki płytki krwi przyczepiają się do ścian uszkodzonego naczynia. W rezultacie komórki sklejają się ze sobą. Z biegiem czasu do powstałej masy dodawane są nici fibrynowe, nowe sklejone komórki i inne elementy.
Na tym tle rośnie skrzep krwi, który osiąga duże rozmiary, co może prowadzić do zablokowania naczynia i zatrzymania krwawienia. Szybkość takiego procesu jest bardzo ważna, ponieważ czasami od tego zależy zachowanie życia ludzkiego.

Na krzepnięcie krwi wpływa wiele czynników. Jednym z nich jest agregacja. W przypadku braku stanów patologicznych pełni ochronną funkcję adaptacyjną.
Cechy agregacji polegają na sklejaniu się komórek tylko w uszkodzonym naczyniu. W takim przypadku proces uważa się za pozytywny.
.jpg)
Są jednak sytuacje, w których zakrzepica jest niepożądana. Na przykład, jeśli zdiagnozowano udar lub zawał mięśnia sercowego.
Wyjaśnia to fakt, że tworzenie się skrzepów krwi uniemożliwia normalny przepływ niezbędnych substancji do ważnych narządów.
W tym przypadku płytki krwi stają po stronie procesów patologicznych. Odchylenia od normy można rozwiązać jedynie za pomocą leków.
Aby odróżnić wskaźniki normalne od nieprawidłowych, konieczne jest przeprowadzenie analizy ilościowej agregacji dodatniej i ujemnej.
Rodzaje
W praktyce medycznej istnieje pewna klasyfikacja agregacji według rodzaju. Obejmują one:

- Umiarkowana agregacja. Diagnozuje się ją głównie w czasie ciąży. Stan ten może być wywołany przez krążenie łożyskowe.
- Spontaniczna agregacja. Do wykrywania nie jest wymagana cewka indukcyjna. Aby wykryć aktywność agregacyjną, krew wlewa się do probówki, którą umieszcza się w specjalnym urządzeniu, gdzie podgrzewa się ją do 37 stopni.
- Indukowana agregacja. W celu przeprowadzenia badania do osocza dodaje się induktory. W tym przypadku następuje agregacja z ADP, kolagenem, ristomycyną i adrenaliną. Metodę tę stosuje się w przypadkach, gdy konieczne jest zdiagnozowanie pewnych patologii płynu krwi.
- Zwiększona agregacja sprzyja tworzeniu się skrzepów krwi. Charakterystycznymi objawami tego stanu patologicznego są drętwienie i obrzęk.
- Zmniejszoną agregację stwierdza się najczęściej w przypadku zaburzeń w funkcjonowaniu układu krążenia. Zmniejszenie liczby płytek krwi powoduje różne krwawienia. Występuje u płci pięknej podczas cyklu menstruacyjnego.
Zarówno zwiększona, jak i zmniejszona agregacja są niebezpieczne dla zdrowia człowieka. Dlatego należy regularnie kontrolować poziom płytek krwi.
Objawy odchyleń od wskaźników
Hiperagregacji towarzyszy zwiększona lepkość krwi i zmniejszenie prędkości jej przepływu, co niekorzystnie wpływa na wszystkie układy i narządy człowieka.
Istnieją jednak stany patologiczne, w których wyraźna agregacja jest zjawiskiem normalnym, co z kolei nie jest uważane za powód do odmowy ciągłego badania wskaźników krzepnięcia.
Do takich chorób należą:
- podwyższone ciśnienie krwi;
- cukrzyca;
- rak;
- patologie naczyniowe.
Niewczesne wykrycie hiperagregacji i brak pomocy może prowadzić do rozwoju zawału serca, udaru mózgu i zakrzepicy żył.
Spadkowi wskaźników agregacji towarzyszy przedłużone krwawienie, w tym krwawienie wewnętrzne, które objawia się powstawaniem krwiaków.
Jaka jest norma
Normy poziomu płytek krwi u osoby dorosłej i dziecka będą nieco inne. Optymalne wartości wskaźników przedstawia poniższa tabela.
Jeśli mówimy o normalnych wartościach agregacji, to będzie to 25-75 proc. W tym przypadku płytki krwi sklejają się ze sobą bez odchyleń i nie stanowią zagrożenia dla organizmu człowieka.
Jakie badania są prowadzone?
Analizator agregacji płytek krwi to pełna morfologia krwi. Istnieją jednak inne badania, które dostarczają dokładniejszych wyników. Główne metody obejmują następujące testy:
.jpg)
- według Lee-White'a;
- koagulogram.
Ich istota polega na tym, że do krwi dodawane są specjalne substancje hamujące agregację.
Składniki te są podobne do substancji zawartych w organizmie człowieka, które powodują powstawanie skrzepów krwi. Takie elementy nazywane są cewkami indukcyjnymi.
Przygotowanie do analizy
Przed wykonaniem analizy należy przejść pewne przygotowanie. Aby wyniki były jak najbardziej dokładne, płyn krwi nie powinien zawierać substancji, które mogłyby mieć na nią negatywny wpływ.
Działania przygotowawcze:
- Na tydzień przed analizą niektóre leki aspiryny są wykluczone, ponieważ ich stosowanie hamuje tworzenie się skrzeplin. W przypadku braku możliwości rezygnacji z tych leków należy powiadomić technika laboratoryjnego prowadzącego badanie.
- Musisz przestać jeść przez 12 godzin. Produkty, szczególnie te o dużej zawartości tłuszczu, również negatywnie wpływają na wyniki.
- Unikaj stresu fizycznego i emocjonalnego.
- W ciągu dnia nie należy pić napojów alkoholowych, kawy, czosnku ani palić papierosów.
Analiza zostaje odroczona, jeśli występuje aktywny proces zapalny.
Przeprowadzanie
Pobieranie krwi odbywa się rano, w godzinach 7-10. Badanie można przeprowadzić wyłącznie na czczo. Dozwolone jest picie wody niegazowanej.
Aby przeprowadzić hemotest, pobiera się płyn krwi z żyły. Do tych celów używa się jednorazowej strzykawki. Następnie materiał umieszcza się w agregometrze zawierającym 4% roztwór cytrynianu sodu. Następnie pojemnik odwraca się kilka razy. Następnie probówka z krwią wysyłana jest do laboratorium w celu dalszych badań.
Dekodowanie wyników
Biorąc pod uwagę substancję, która została użyta podczas badania, analiza zostaje rozszyfrowana. W tym celu uzyskane wskaźniki porównuje się z wartościami normalnymi, które przedstawiono poniżej.
Jeśli zaobserwowano wzrost w stosunku do normy, rozpoznaje się hiperagregację. Może wystąpić w stanach patologicznych, takich jak:
.jpg)
- białaczka;
- patologie przewodu żołądkowo-jelitowego lub nerek;
- miażdżyca;
- cukrzyca;
- wysokie ciśnienie krwi;
- posocznica;
- limfogranulomatoza.
Jeśli odchylenia są w dół, diagnozuje się hipoagregację. Może to być spowodowane patologiami krwi lub leczeniem lekami przeciwpłytkowymi.
Procent pokazuje poziom przepuszczalności światła plazmy po dodaniu do niej substancji induktora. Jeśli liczba płytek krwi jest niska, wskaźnik ten wynosi 100 procent, a jeśli jest wysoka, wynosi zero.
Cechy agregacji u kobiet w ciąży
W czasie ciąży dopuszczalne są odchylenia od normy, które w tym okresie wahają się od 30 do 60 procent.
Dezagregację można zaobserwować w przypadku braku płytek krwi, a także w przypadku zmiany ich składu jakościowego, co objawia się krwawieniem i siniakami.
Zwiększona agregacja występuje podczas zatrucia, gdy pacjent doświadcza dużej utraty płynów w wyniku wymiotów lub biegunki. Wzrost stężenia we krwi powoduje zwiększone tworzenie się skrzepów krwi. Grozi to wczesnym poronieniem.
Jak normalizować wartości?
W przypadku zdiagnozowania zaburzenia krzepnięcia krwi konieczne jest natychmiastowe podjęcie działań w celu wyeliminowania stanu patologicznego. Zwiększenie agregacji może prowadzić do zakrzepicy, a zmniejszenie może prowadzić do ciężkiego i niebezpiecznego krwawienia.
Na początkowych etapach rozwoju hiperagregacji specjaliści przepisują leki, które mogą rozrzedzać krew. Zwykła aspiryna poradzi sobie z tym zadaniem.
Na podstawie wyników dodatkowego badania często przepisuje się:
.jpg)
- środki przeciwbólowe;
- blokada nowokainy;
- leki promujące rozszerzenie naczyń;
- antykoagulanty, które zapobiegają szybkiemu krzepnięciu.
Czasami tradycyjne metody są nie mniej skuteczne. Warto pamiętać, że takie leczenie należy uzgodnić z lekarzem prowadzącym.
Wśród sprawdzonych przepisów są następujące:
- Łyżkę słodkiej koniczyny zalać 200 ml przegotowanej wody i pozostawić na 30 minut. Przygotowaną mieszankę stosować dziennie w kilku dawkach. Przebieg terapii wynosi jeden miesiąc.
- Zaparzyć półtora litra wrzącej wody z równych ilości (łyżeczka) imbiru i zielonej herbaty. Dodaj szczyptę cynamonu. Pozostaw na kwadrans i zażywaj przez cały dzień.
- Codziennie pij świeżo wyciśnięty sok pomarańczowy. Można mieszać w równych częściach z dynią.
Ważne jest także dbanie o prawidłowe odżywianie. Dieta powinna zawierać:
- cytrus;
- ożywić;
- czosnek;
- warzywa czerwone i zielone;
- owoce morza.
Jeśli masz słabą krzepliwość krwi, nie powinieneś brać leków. które rozrzedzają płyn krwi. Jeśli przebieg procesu nabrał zaawansowanej formy, wówczas działania terapeutyczne przeprowadzane są wyłącznie w warunkach stacjonarnych.
Przepisywane są następujące leki:

- Emosint;
- Kwas aminokapronowy i traneksamowy;
- zastrzyk ATP;
Dieta musi uwzględniać kaszę gryczaną, jajka, buraki i marchewkę, granat, wątrobę wołową i czerwone mięso.
Aby utrzymać krew w normalnym stanie, konieczne jest ścisłe przestrzeganie reżimu picia. Za normę uważa się co najmniej półtora litra czystej wody dziennie. Jedzenie powinno być świeże i zbilansowane.
Przestrzeganie zasad żywienia to zapobieganie wielu chorobom organizmu człowieka. Równie ważną rolę odgrywa aktywność fizyczna. Pomagają nie tylko wzmocnić organizm, ale także normalizować wszystkie procesy wewnętrzne.
Dzięki terminowej diagnozie odchyleń wskaźników agregacji można zapobiec wielu chorobom i powikłaniom. Należy regularnie monitorować poziom agregacji płytek krwi.
Płytki krwi to małe komórki krwi odpowiedzialne za krzepnięcie krwi. Pomagają zatrzymać utratę krwi w przypadku wystąpienia krwawienia.
Kiedy pojawia się rana, płytki krwi przemieszczają się do uszkodzonego obszaru. Tutaj są przymocowane do ściany uszkodzonego naczynia, w wyniku czego krwawienie ustaje. Proces ten nazywany jest agregacją płytek krwi.
Agregacja płytek krwi to proces, w którym komórki krwi przylegają do siebie i mocują je na ścianie uszkodzonego naczynia. To zatrzymuje krwawienie. Jednak taki proces może być również niebezpieczny dla organizmu. W takim przypadku tworzy się skrzep krwi, który w pewnych okolicznościach może wywołać zawał serca i udar. Może się to zdarzyć, jeśli płytki krwi są nadaktywne i agregacja następuje zbyt szybko.
Ponadto powolny proces również nie obiecuje niczego dobrego dla organizmu. W takim przypadku może wystąpić słaba krzepliwość krwi z powodu powolnej adhezji płytek krwi. Ta patologia powoduje anemię. Jeśli krzepnięcie krwi jest słabe, trudno jest zatrzymać krwawienie, co może prowadzić do problemów zdrowotnych, a nawet śmierci. Aby temu zapobiec, należy monitorować poziom płytek krwi we krwi i ich zdolność do sklejania się.
Proces agregacji płytek krwi w czasie ciąży
 Niezwykle ważne jest, aby agregacja płytek krwi w czasie ciąży przebiegała prawidłowo. Jeśli proces zachodzi zbyt wolno, wówczas podczas porodu lub w okresie poporodowym może wystąpić krwawienie z macicy, co może prowadzić do śmierci kobiety. Ponadto, jeśli w czasie ciąży agregacja płytek krwi nastąpi szybko, mogą powstać skrzepy krwi, co w czasie ciąży może prowadzić do jej przerwania na każdym etapie.
Niezwykle ważne jest, aby agregacja płytek krwi w czasie ciąży przebiegała prawidłowo. Jeśli proces zachodzi zbyt wolno, wówczas podczas porodu lub w okresie poporodowym może wystąpić krwawienie z macicy, co może prowadzić do śmierci kobiety. Ponadto, jeśli w czasie ciąży agregacja płytek krwi nastąpi szybko, mogą powstać skrzepy krwi, co w czasie ciąży może prowadzić do jej przerwania na każdym etapie.
Możesz uniknąć tej sytuacji, jeśli zaplanujesz ciążę i zadbasz o swoje zdrowie z wyprzedzeniem. Konieczne jest, jeszcze przed poczęciem, sprawdzenie, w jakim stanie są płytki krwi i, jeśli to konieczne, podjęcie działań w celu poprawy sytuacji. Jeśli ciąża nie była planowana, można uniknąć patologii agregacji, rejestrując się na wczesnym etapie. Następnie lekarz przepisze niezbędne badania i pomoże pozbyć się patologicznego stanu płytek krwi, jeśli zostanie wykryty.
Normalna liczba płytek krwi we krwi
Aby poznać stan poziomu płytek krwi, musisz mieć pojęcie o ich normalnym poziomie.
Jeśli mówimy o współczynniku agregacji, to wynosi on 25-75%. W tym przypadku proces sklejania płytek krwi przebiega prawidłowo i nie stwarza żadnego zagrożenia dla zdrowia.
Jeśli zostaną zaobserwowane odchylenia od normy, zaleca się odpowiednie leczenie, aby pomóc poprawić sytuację.Badanie krwi na agregację płytek krwi
Badanie krwi zwane agregacją indukowaną pomaga zbadać stan płytek krwi. W tym przypadku krew pobierana jest z żyły pacjenta i mieszana ze specjalnymi substancjami. Środki takie mają skład zbliżony do składu komórek organizmu biorących udział w procesie agregacji. Jako induktory najczęściej przyjmuje się następujące substancje:
- difosforan adenozyny (ADP);
- kolagen;
- serotonina;
- rystocetyna;
- adrenalina;
- kwas arachidonowy.
Najczęściej wykonywaną procedurą jest agregacja płytek krwi za pomocą ADP. Aby wykonać badanie, pobierane jest specjalne urządzenie. Nazywa się to analizatorem agregacji płytek krwi. Za jego pomocą fale świetlne przepuszczane są przez krew zanim zacznie ona krzepnąć oraz po zakończeniu tego procesu. Następnie oceniany jest wynik.
Przygotowanie do testów
 Aby mieć pewność, że wynik jest jak najbardziej dokładny, należy przestrzegać następujących zasad wykonywania badania krwi:
Aby mieć pewność, że wynik jest jak najbardziej dokładny, należy przestrzegać następujących zasad wykonywania badania krwi:
- Badanie przeprowadza się na czczo. W takim przypadku należy przerwać jedzenie na 12 godzin przed badaniem. W takim przypadku możesz pić czystą wodę niegazowaną.
- Na 7 dni przed badaniem należy przerwać leczenie niektórymi lekami. Jeśli nie jest to możliwe, należy poinformować lekarza przeprowadzającego analizę.
- Na kilka dni przed analizą należy unikać stresujących sytuacji i aktywności fizycznej.
- Należy przestać pić kawę, palić, pić alkohol i czosnek z 24-godzinnym wyprzedzeniem.
- Nie można przeprowadzić badań, jeśli w organizmie występuje proces zapalny.
Wskazania do analizy
Możesz od czasu do czasu z własnej inicjatywy wykonać badanie agregacji płytek krwi. Interpretacja wyniku analizy agregacji indukowanej
Interpretacja wskaźników zależy od środków, jakimi przeprowadzono badanie. W tym celu dane porównuje się z normą.
Jeśli wyniki odbiegają od normy w górę, rozpoznaje się zwiększoną agregację płytek krwi. Warunek ten występuje, gdy: 
- wysokie ciśnienie krwi;
- miażdżyca;
- białaczka;
- cukrzyca;
- choroby onkologiczne przewodu żołądkowo-jelitowego lub nerek;
- limfogranulomatoza;
- posocznica;
- chirurgiczne usunięcie śledziony.
Zwiększona agregacja płytek krwi może prowadzić do zawału serca, udaru mózgu, zakrzepicy i śmierci z powodu zablokowania naczynia przez skrzep krwi.
Jeżeli wyniki odbiegają od normy w kierunku spadku, wówczas rozpoznaje się zmniejszoną agregację zakrzepicy. Jest to spowodowane:
- choroby krwi;
- trombocytopatia;
- stosowanie leków przeciwpłytkowych.
Przy zmniejszonej agregacji naczynia stają się kruche. Ponadto proces tamowania krwawienia jest trudny, co może spowodować śmierć.
Środki redukujące proces agregacji
Niektóre środki hamują proces agregacji. Leki te obejmują leki przeciwpłytkowe. Inhibitory agregacji płytek krwi obejmują leki takie jak kwas acetylosalicylowy, ibustryna, mikrystyna i inne. Takie leki są przepisywane w celu leczenia niektórych chorób. Jeśli jednak proces agregacji znacznie odbiega od normy, leki hamujące należy zastąpić innymi środkami, które nie prowadzą do takich konsekwencji. Jeśli nie jest to możliwe, lekarz może przepisać specjalne leki sprzyjające agregacji.
Zarówno zwiększona, jak i zmniejszona agregacja może prowadzić do tragicznych konsekwencji. Dlatego nie można ignorować takich sytuacji. W takich przypadkach należy skonsultować się z lekarzem, który zaleci odpowiednie leczenie.Również polecamy
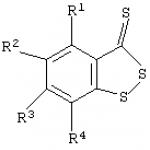 Leki wpływające na hemostazę płytek naczyniowych Agregację płytek krwi hamuje bloker Ca2
Leki wpływające na hemostazę płytek naczyniowych Agregację płytek krwi hamuje bloker Ca2
 Czym karmić mózg Mediatory neuroprzekaźnika pobudzającego adrenalinę w układzie nerwowym
Czym karmić mózg Mediatory neuroprzekaźnika pobudzającego adrenalinę w układzie nerwowym
 Zwiększony poziom fosfatazy alkalicznej
Zwiększony poziom fosfatazy alkalicznej
 Leki uzależniające Objawy uzależnienia od tramadolu
Leki uzależniające Objawy uzależnienia od tramadolu
 Leki przeciwhistaminowe najnowszej generacji
Leki przeciwhistaminowe najnowszej generacji
 Lek Finasteryd na łysienie Finasteryd na recenzje włosów
Lek Finasteryd na łysienie Finasteryd na recenzje włosów