Caractéristiques générales des éléments du groupe V-A. Éléments P du groupe V Valence maximale des éléments du groupe 5 du sous-groupe principal
Similitudes des éléments :
Structure identique de la couche électronique externe des atomes ns 2 np 3 ;
Éléments P ;
Village supérieur O. égal à +5 ;
Inférieur s. O. égal à -3 (inhabituel pour Sb et Bi).
Pour les éléments du sous-groupe principal du groupe V, on utilise parfois le nom de groupe « pnictogènes », introduit par analogie avec les termes « halogènes » et « chalcogènes » et dérivé des symboles des éléments phosphore P et azote N.
États de Valence des atomes
Pour les atomes P, As, Sb, Bi, 2 états de valence sont possibles :
Base ns 2 np 3
Horny ns 1 np 3 ème 1
La différence entre l'azote et les autres éléments du sous-groupe
1. En raison de l'absence d'orbitales D dans la couche électronique externe de l'atome d'azote, le nombre de liaisons covalentes formées par l'atome d'azote selon le mécanisme d'échange ne peut pas être supérieur à 3.
2. La présence d'une paire d'électrons libres au sous-niveau 2s de l'atome d'azote permet de former une liaison covalente via le mécanisme donneur-accepteur. Ainsi, la valence la plus élevée de N est IV.
3. Dans les composés avec de l'oxygène, l'azote présente les états d'oxydation +1, +2, +3, +4, +5.
Changement vertical des propriétés des éléments et des substances qu'ils forment
Contrairement aux halogènes et aux chalcogènes, dans le sous-groupe principal du groupe V, il y a un changement plus spectaculaire dans les propriétés des éléments et des substances simples qu'ils forment à mesure que la charge du noyau et le rayon des atomes augmentent :
Changement vertical des propriétés des éléments et des substances simples qu'ils forment |
|||||
non-métaux |
métal avec quelques signes de non-métallicité |
||||
Oxydes et hydroxydes |
L'azote et ses composés oxygénés sont considérés séparément en raison d'un certain nombre de différences. |
||||
E 2 O 3 et hydroxydes correspondants |
Oxyde d'acide P 2 O 3 (P 4 O 6) |
Comme oxyde acide 2 O 3 avec des signes d'amphotéricité |
Oxyde amphotère Sb 2 O 3 |
Oxyde basique Bi 2 O 3 |
|
E 2 O 5 et hydroxydes correspondants |
Oxyde d'acide P 2 O 5 (P 4 O 10) |
Comme oxyde d'acide 2 O 5 |
Oxyde d'acide Sb 2 O 5 |
Bi 2 O 5 amphotère fragile |
|
HPO 3 (H 3 PO 4) |
|||||
acides faibles |
|||||
Les propriétés acides s'affaiblissent |
|||||
Les propriétés de base sont améliorées |
|||||
Composés hydrogènes EN 3
Les éléments du sous-groupe principal du groupe V forment des composés volatils avec l'hydrogène, qui sont des gaz toxiques aux odeurs caractéristiques. Ce sont de puissants agents réducteurs. Contrairement aux composés hydrogènes des non-métaux des groupes VII et VI, ils ne forment pas d'ions H + dans les solutions aqueuses, c'est-à-dire qu'ils ne présentent pas de propriétés acides.
CARACTÉRISTIQUES GÉNÉRALES DU 5ÈME GROUPE D'ÉLÉMENTS.
Le sous-groupe principal V 0 01 F du tableau périodique comprend l'azote, le phosphore, l'arsenic, l'antimoine et le bismuth. Ces éléments, possédant cinq électrons dans la couche externe de l'atome, sont caractérisés en général
comme les non-métaux. Cependant, leur capacité à ajouter des électrons est bien moins prononcée que celle des éléments correspondants des groupes VI et VII. En raison de la présence de cinq électrons externes, l'oxydation positive la plus élevée des éléments de ce sous-groupe est de -5 et l'oxydation négative est de 3. En raison de l'électronégativité relativement plus faible, la liaison des éléments en question avec l'hydrogène est moins polaire que la liaison avec l'hydrogène des éléments des groupes VI et VII. Par conséquent, les composés hydrogène de ces éléments n’éliminent pas les ions hydrogène H dans une solution aqueuse et n’ont donc pas de propriétés acides. Les propriétés physiques et chimiques des éléments du sous-groupe azote changent avec
0 0 1 Finaugmentation du numéro de série dans la même séquence que celle observée dans
0 0 1 Pour les groupes précédemment considérés, Mais comme les propriétés non métalliques sont exprimées en
plus faible que celle de l'oxygène et surtout du fluor, alors l'affaiblissement de ces propriétés lors du passage à 0 0 1 F des éléments suivants entraîne l'apparition et l'augmentation de propriétés métalliques.
Ces dernières sont déjà visibles dans l'arsenic, l'antimoine a les deux propriétés à peu près égales et dans le bismuth, les propriétés métalliques prédominent sur les propriétés non métalliques. DESCRIPTION DES ÉLÉMENTS. AZOTE(du grec ázōos - sans vie, lat. Nitrogenium), N, élément chimique V
groupes du système périodique de Mendeleev, numéro atomique 7, masse atomique 14,0067 ; gaz incolore, inodore et insipide. Référence historique. Les composés azotés - salpêtre, acide nitrique, ammoniac - ont été
connue bien avant la production d’azote à l’état libre. En 1772, D. Rutherford, brûlant du phosphore et d'autres substances dans une cloche en verre, montra que le gaz restant après la combustion, qu'il appelait « air suffocant », ne supportait pas la respiration et la combustion. En 1787, A. Lavoisier établit que les gaz « vitaux » et « asphyxiants » qui composent l'air sont des substances simples et propose le nom d'« azote ». En 1784, G. Cavendish montra que l'azote fait partie du salpêtre ; C'est de là que vient le nom latin azote (du latin tardif nitrum - salpêtre et du grec gennao - j'accouche, je produis), proposé en 1790 par J. A. Chaptal. Au début du 19ème siècle. L'inertie chimique de l'azote à l'état libre et son rôle exclusif dans les composés avec d'autres éléments comme l'azote lié ont été clarifiés. Depuis lors, la « liaison » de l’azote de l’air est devenue l’un des problèmes techniques les plus importants de la chimie. Prévalence dans la nature. L'azote est l'un des éléments les plus courants sur
La Terre et sa masse (environ 4´1015 T) est concentré à l’état libre dans l’atmosphère. Dans l'air, l'azote libre (sous forme de molécules N2) est de 78,09 % en volume (soit 75,6 % en masse), sans compter ses impuretés mineures sous forme d'ammoniac et d'oxydes. La teneur moyenne en azote dans la lithosphère est de 1,9´10-3 % en masse. Composés azotés naturels. - du chlorure d'ammonium NH4Cl et divers nitrates (voir.
Salpêtre.) Les grandes accumulations de salpêtre sont caractéristiques d'un climat désertique sec (Chili, Asie centrale). Pendant longtemps, le nitrate a été le principal fournisseur d'azote pour l'industrie (aujourd'hui, la synthèse industrielle de l'ammoniac à partir de l'azote de l'air et de l'hydrogène est primordiale pour la fixation de l'azote). De petites quantités d'azote fixe se trouvent dans le charbon (1 à 2,5 %) et le pétrole (0,02 à 1,5 %), ainsi que dans les eaux des rivières, des mers et des océans. L'azote s'accumule dans les sols (0,1 %) et dans les organismes vivants (0,3 %).
Bien que le nom « azote » signifie « qui ne maintient pas la vie », il s’agit en réalité d’un élément essentiel à la vie. Les protéines animales et humaines contiennent 16 à 17 % d'azote. Dans les organismes des animaux carnivores, les protéines se forment en raison de la consommation de substances protéiques présentes dans les organismes des animaux herbivores et dans les plantes. Les plantes synthétisent des protéines en assimilant les substances azotées contenues dans le sol, principalement inorganiques. Des quantités importantes d'azote pénètrent dans le sol grâce à des micro-organismes fixateurs d'azote capables de convertir l'azote libre de l'air en composés azotés. Le cycle de l'azote se produit dans la nature, dans lequel le rôle principal est joué par
micro-organismes - nitrophisation, dénitrophie, fixation de l'azote, etc. Cependant, en raison de l'extraction d'énormes quantités d'azote fixe du sol par les plantes (en particulier dans le cas d'une agriculture intensive), les sols s'épuisent en azote. La carence en azote est typique de l’agriculture dans presque tous les pays ; une carence en azote est également observée dans l’élevage (« famine en protéines »). Sur les sols pauvres en azote disponible, les plantes se développent mal. Les engrais azotés et l’alimentation protéique des animaux constituent les moyens les plus importants pour stimuler l’agriculture. Les activités économiques humaines perturbent le cycle de l’azote. Ainsi, la combustion de carburant enrichit l’atmosphère en azote et les usines produisant des engrais captent l’azote de l’air. Le transport d’engrais et de produits agricoles redistribue l’azote à la surface de la terre. L'azote est le quatrième élément le plus abondant dans le système solaire (après l'hydrogène,
hélium et oxygène). Isotopes, atome, molécule. L'azote naturel est constitué de deux isotopes stables : 14N
(99,635%) et 15N (0,365%). L'isotope 15N est utilisé dans la recherche chimique et biochimique comme atome marqué. Parmi les isotopes radioactifs artificiels de l'azote, le 13N a la demi-vie la plus longue (T1/2 - 10,08 min), le reste est de très courte durée. Dans les couches supérieures de l'atmosphère, sous l'influence des neutrons du rayonnement cosmique, le 14N se transforme en isotope radioactif du carbone 14C. Ce processus est également utilisé dans les réactions nucléaires pour produire du 14C. La couche électronique externe de l’atome d’azote. se compose de 5 électrons (un doublet libre et trois non appariés - configuration 2 s 22p 3). Le plus souvent de l'azote. dans les composés, il est 3-covalent en raison d'électrons non appariés (comme dans l'ammoniac NH3). La présence d’un doublet libre d’électrons peut conduire à la formation d’une autre liaison covalente, et l’azote devient 4-covalent (comme dans l’ion ammonium NH4+). Les états d'oxydation de l'azote varient de +5 (dans N205) à -3 (dans NH3). Dans des conditions normales, à l'état libre, l'azote forme une molécule N2, où les atomes N sont reliés par trois liaisons covalentes. La molécule d'azote est très stable : son énergie de dissociation en atomes est de 942,9 kJ/mole (225,2 kcal/mole), donc même avec t degré de dissociation de l'azote d'environ 3 300 °C. n'est que d'environ 0,1 %. Proprietes physiques et chimiques. L'azote est légèrement plus léger que l'air ; densité 1.2506 kg/
m3(à 0°C et 101325 m/m2 ou 760 mmHg Art.), s'il vous plaît-209,86°C, faire bouillir-195,8 ?C. A. se liquéfie difficilement : sa température critique est assez basse (-147,1°C), et sa pression critique est élevée 3,39 Mn/m2 (34,6 kgf/cm2); densité de l'azote liquide 808 kg(m3. L'azote est moins soluble dans l'eau que l'oxygène : à 0°C en 1 m3 H2O dissout 23,3 g azote. L'azote est plus soluble dans certains hydrocarbures que dans l'eau. Uniquement avec des métaux actifs tels que le lithium, le calcium, le magnésium, l'azote
interagit lorsqu’il est chauffé à des températures relativement basses. L'azote réagit avec la plupart des autres éléments à haute température et en présence de catalyseurs. Les composés de l'azote avec l'oxygène N2O, NO, N2O3, NO2 et N2O5 ont été bien étudiés. À partir de ceux-ci, lors de l'interaction directe des éléments (4 000 °C), de l'oxyde de NO se forme qui, lors du refroidissement, s'oxyde facilement en dioxyde de NO2. Dans l'air, des oxydes d'azote se forment lors des rejets atmosphériques. Ils peuvent également être obtenus en exposant un mélange d'azote et d'oxygène à des rayonnements ionisants. Lorsque le N2O3 azoté et
Les anhydrides nitriques N2O5 produisent respectivement de l'acide nitreux HNO2 et de l'acide nitrique HNO3, formant des sels - nitrites et nitrates. L'azote se combine à l'hydrogène uniquement à haute température et en présence de catalyseurs, et de l'ammoniac NH3 se forme. En plus de l'ammoniac, de nombreux autres composés d'azote et d'hydrogène sont connus, par exemple l'hydrazine H2N-NH2, le diimide HN-NH, l'acide hydronitrique HN3(H-N-NºN), l'octazone N8H14, etc. La plupart des composés azotés et hydrogènes sont isolés uniquement sous forme de dérivés organiques. L'azote n'interagit pas directement avec les halogènes, donc tous les halogénures d'azote ne sont obtenus qu'indirectement, par exemple le fluorure d'azote NF3- lorsque le fluor réagit avec l'ammoniac. En règle générale, les halogénures d'azote sont des composés peu stables (à l'exception du NF3) ; Les oxyhalogénures d'azote sont plus stables - NOF, NOCI, NOBr, N02F et NO2CI. Il n’existe pas non plus de combinaison directe d’azote et de soufre ; le soufre azoté N4S4 est obtenu à la suite de la réaction du soufre liquide avec l'ammoniac. Lorsque le coke chaud réagit avec l'azote, du cyanogène (CN) se forme.;. En chauffant de l'azote avec de l'acétylène C2H2 à 1 500 °C, du cyanure d'hydrogène HCN peut être obtenu. L'interaction de l'azote avec les métaux à haute température conduit à la formation de nitrures (par exemple Mg3N2). Lorsque l'azote ordinaire est exposé à des décharges électriques [pression 130 - 270 m/m2(1- 2
mmHg)] ou lors de la décomposition des nitrures de B, Ti, Mg et Ca, ainsi que lors de décharges électriques dans l'air, de l'azote actif peut se former, qui est un mélange de molécules et d'atomes d'azote avec une réserve d'énergie accrue. Contrairement à l’azote moléculaire, l’azote actif interagit de manière très énergétique avec l’oxygène, l’hydrogène, les vapeurs de soufre, le phosphore et certains métaux. L'azote fait partie de nombreux composés organiques importants (amines,
acides aminés, composés nitro, etc.). Réception et demande. En laboratoire, l'azote peut facilement être obtenu par
chauffer une solution concentrée de nitrite d'ammonium : NH4NO2 - N2 + 2H2O. Le procédé technique de production d'azote repose sur la séparation de l'air pré-liquéfié, qui est ensuite soumis à une distillation. La majeure partie de l’azote libre extrait est utilisée à des fins industrielles.
production d'ammoniac, qui est ensuite transformé en quantités importantes en acide nitrique, engrais, explosifs, etc. Outre la synthèse directe de l'ammoniac à partir d'éléments, la méthode au cyanamide s'est développée en 1905, basée sur le fait qu'à 1000 °C du carbure de calcium (obtenu en chauffant un mélange de chaux et de charbon dans un four électrique) réagit avec l'azote libre : CaC2 + N2 = CaCN2 + C. Le cyanamide calcique obtenu se décompose sous l'action de la vapeur d'eau surchauffée, libérant de l'ammoniac :
CaCN+3H2O=CaCO3+2NH3.
L'azote libre est utilisé dans de nombreuses industries : comme milieu inerte dans divers procédés chimiques et métallurgiques, pour remplir l'espace libre des thermomètres à mercure, lors du pompage de liquides inflammables, etc. L'azote liquide est utilisé dans diverses unités de réfrigération. Il est stocké et transporté dans des récipients Dewar en acier, l'azote gazeux sous forme comprimée - dans des bouteilles. De nombreux composés azotés sont largement utilisés ; la production d'azote fixe a commencé à se développer rapidement après la Première Guerre mondiale et a aujourd'hui atteint des proportions énormes.
Phosphore(lat. Phosphore), P, élément chimique du groupe V du système périodique de Mendeleev, numéro atomique 15, masse atomique 30,97376, non métallique. Le phosphore naturel est constitué d'un isotope stable, le 31P ; six isotopes radioactifs artificiels ont été obtenus : 28P ( T1/2 - 6,27 seconde), 29P ( T1/2 - 4,45 s) ; 30P ( T1/2 - 2,55 min), 31P ( T1/2
14,22 jours), 32P ( T1/2 - 25 jours), 33P ( T1/2 - 12,5 seconde). 32P a la plus grande valeur , possédant une énergie de rayonnement b importante et utilisé dans la recherche chimique et biochimique en tant qu'atome marqué. Référence historique. Selon certaines données de la littérature, la méthode d'obtention
Le phosphore était déjà connu des Arabes. alchimistes du XIIe siècle. Mais la date généralement acceptée pour la découverte du phosphore est considérée comme 1669, lorsque H. Brand (Allemagne), en calcinant le résidu sec de l'évaporation de l'urine avec du sable et en le distillant ultérieurement sans accès à l'air, obtint une substance qui brille dans le sombre, d’abord appelé « feu froid », puis phosphore du grec. phosphoros - lumineux. Bientôt, la méthode d'obtention du phosphore lui fut connue. chimistes - I. Kraft, I. Kunkel ; en 1682, cette méthode fut publiée. En 1743, A. S. Marggraf développa la méthode suivante pour produire du phosphore : un mélange de chlorure de plomb et d'urine était évaporé à sec et chauffé jusqu'à ce que le dégagement de produits volatils cesse ; le résidu était mélangé avec du charbon de bois en poudre et distillé dans une cornue en argile ; Vapeur de phosphore condensée dans le récepteur avec de l'eau. La méthode la plus simple pour obtenir du phosphore par calcination de cendres d'os avec du charbon n'a été proposée qu'en 1771 par K. Scheele. La nature élémentaire du phosphore a été établie par A. Lavoisier. Dans la 2ème moitié du 19ème siècle. la production industrielle de phosphore à partir de phosphorites dans des fours à cornue est apparue ; au début du 20e siècle. ils ont été remplacés par des fours électriques. Répartition dans la nature. Teneur moyenne en phosphore dans la croûte terrestre (clark) -
9,3 x 10-2 % en poids ; dans les roches moyennes 1,6×10-1, dans les roches basiques 1,4×10-1, moins dans les granites et autres roches ignées acides - 7×10-2 et encore moins dans les roches ultrabasiques (manteau) - 1, 7×10-2% ; dans les roches sédimentaires de 1,7×10-2 (grès) à 4×10-2% (roches carbonatées). Le phosphore participe aux processus magmatiques et migre vigoureusement dans la biosphère. Les deux processus sont associés à ses grandes accumulations, formant des gisements industriels d'apatites et de phosphorites. . Le phosphore est un élément biogénique extrêmement important ; il est accumulé par de nombreux organismes. De nombreux processus de concentration du phosphore dans la croûte terrestre sont associés à une migration biogénique. Le phosphore est facilement précipité des eaux sous forme de minéraux insolubles ou capturé par la matière vivante. Par conséquent, l’eau de mer ne contient que 7 × 10 à 6 % de phosphore. Environ 180 minéraux phosphorés sont connus, principalement divers phosphates, parmi lesquels les phosphates de calcium sont les plus courants. . Propriétés physiques. Le phosphore élémentaire existe sous la forme de plusieurs
modifications allotropiques dont les principales sont le blanc, le rouge et le noir. Le phosphore blanc est une substance cireuse et transparente avec une odeur caractéristique, formée par la condensation de vapeurs de phosphore. Phosphore blanc en présence d'impuretés - traces de phosphore rouge, d'arsenic, de fer, etc. - peint en jaune, donc phosphore blanc commercial. appelé jaune. Il existe deux formes de phosphore blanc : la forme a et la forme b. a- la modification représente les cristaux du système cubique ( UN- 18,5); densité 1.828 g/cm3, t pl 44,1 °C, t kip 280,5 °C, chaleur de fusion 2,5 kJ/mole P4 (0,6 kcal/mole P4), chaleur d'évaporation 58,6 kJ/mole P4(14,0 kcal/mole P4) , pression de vapeur à 25 °C 5,7 m/m2 (0,043 mmHg Art.). Le coefficient de dilatation linéaire dans la plage de température de 0 à 44 °C est de 12,4 × 10-4 , conductivité thermique 0,56 Mar/(m×K) à 25 °C. Les propriétés électriques du phosphore blanc. proche des diélectriques : bande interdite environ 2,1 ev, résistivité électrique 1,54×1011 ohm× cm, susceptibilité magnétique diamagnétique et spécifique - 0,86×10-6. Dureté Brinell 6 Mn/m2 (0,6 kgf/mm2). La forme a du phosphore blanc se dissout bien dans le sulfure de carbone, moins bien dans l'ammoniac liquide, le benzène, le tétrachlorure de carbone, etc. À - 76,9 °C et une pression de 0,1 Mn/m2(1kgf/cm2) la forme a se transforme en forme b à basse température (densité 1,88 g/cm3). Avec une pression augmentant à 1200 Mn/m2(12 mille kgf/cm2) la transition se produit à 64,5 °C. Forme B - cristaux à biréfringence, leur structure n'est pas définitivement établie. Le phosphore blanc est toxique : dans l'air à une température d'environ 40 °C
s'enflamme automatiquement, il doit donc être stocké sous l'eau (solubilité dans l'eau à 25 °C 3,3 × 10-4 %) . En chauffant le phosphore blanc sans accès à l'air à 250-300 °C pendant plusieurs heures, on obtient du phosphore rouge. La transition est exothermique, accélérée par les rayons ultraviolets, ainsi que par les impuretés (iode, sodium, sélénium). Le phosphore rouge commercial ordinaire est presque complètement amorphe ; a une couleur allant du brun foncé au violet. Avec un chauffage prolongé, il peut se transformer de manière irréversible en l'une des formes cristallines (triclinique, cubique, etc.) aux propriétés différentes : densité de 2,0 à 2,4. g/cm 3, t pl de 585 à 610 °C à une pression de plusieurs dizaines d'atmosphères, température de sublimation de 416 à 423 °C, résistivité électrique de 109 à 1014 ohm× cm. Le phosphore rouge ne s'enflamme pas spontanément dans l'air ; jusqu'à une température de 240-250 °C, mais s'enflamme spontanément lors d'un frottement ou d'un impact ; insoluble dans l'eau, ainsi que dans le benzène, le sulfure de carbone, etc., soluble dans le tribromure de phosphore. À la température de sublimation, le phosphore rouge se transforme en vapeur qui, lors du refroidissement, produit principalement du phosphore blanc. Lorsque le phosphore blanc est chauffé à 200-220 °C sous pression (1,2-1,7) × 103 Mn/m 2
[(12-17)×103 kgf/cm 2] du phosphore noir se forme. Cette transformation peut s'effectuer sans pression, mais en présence de mercure et d'un peu de cristaux de phosphore noir (graine) à 370 °C pendant 8 jours Le phosphore noir est un cristal de structure orthorhombique ( UN -3,31 , b - 4,38 , Avec-10.50), le treillis est constitué de couches fibreuses contenant du phosphore caractéristique. disposition pyramidale des atomes, densité 2,69 g/cm3, t pl environ 1000 °C sous pression 1,8×103 Mn/m2(18×103 kgf/cm2). En apparence, le phosphore noir est similaire au graphite ; semi-conducteur : bande interdite 0,33 evà 25 °C ; a une résistivité électrique spécifique de 1,5 ohm× cm, le coefficient de température de la résistance électrique est de 0,0077, la susceptibilité magnétique diamagnétique et spécifique est de 0,27×10-6. Lorsqu'il est chauffé à 560-580 °C sous la pression de ses propres vapeurs, il se transforme en phosphore rouge. Le phosphore noir est peu actif et difficile à enflammer lorsqu'il est allumé, il peut donc être traité en toute sécurité dans l'air. Le rayon atomique du phosphore est de 1,34, rayons ioniques : P5 + 0,35, P3 + 0,44, P3- 1,86.
Les atomes de phosphore se combinent pour former des molécules diatomiques (P2), tétraatomiques (P4) et polymères. Les molécules polymères les plus stables dans des conditions normales sont celles contenant de longues chaînes de tétraèdres P4 interconnectés. Sous forme liquide, solide (phosphore blanc) et vapeur en dessous de 800 °C, le phosphore est constitué de molécules P4. À des températures supérieures à 800 °C, les molécules P4 se dissocient en P2 , qui, à leur tour, se désintègrent en atomes à des températures supérieures à 2 000 °C. Seul le phosphore blanc est constitué de molécules P4, toutes les autres modifications sont des polymères. Propriétés chimiques. Configuration des électrons externes de l'atome de phosphore 3 s2 3p3, V
composés, les états d'oxydation les plus caractéristiques sont + 5, + 3 et - 3. Comme l'azote, le phosphore présent dans les composés est principalement covalent. Il existe très peu de composés ioniques comme les phosphures Na3P et Ca3P2. Contrairement à l'azote, le phosphore possède 3 d- des orbitales aux énergies plutôt basses, ce qui conduit à la possibilité d'augmenter le nombre de coordination et de former des liens donneur-accepteur. Le phosphore est chimiquement actif, le phosphore blanc a la plus grande activité ;
le phosphore rouge et noir est beaucoup plus passif dans les réactions chimiques. L'oxydation du phosphore blanc se produit via un mécanisme de réaction en chaîne. L'oxydation du phosphore s'accompagne généralement d'une chimiluminescence. Lorsque le phosphore brûle en présence d'un excès d'oxygène, il se forme du pentoxyde P4O10 (ou P2O5) en cas de carence, principalement du trioxyde P4O6 (ou P2O3). L'existence de P4O7, P4O8 dans les vapeurs a été prouvée spectroscopiquement , P2O6, PO et autres oxydes de phosphore. Le pentoxyde de phosphore est produit à l'échelle industrielle en brûlant du phosphore élémentaire dans un excès d'air sec. L'hydratation ultérieure du P4O10 conduit à la production d'ortho-(H3PO4) et de poly-(H n+2 P. n O3 P.+ 1) acides phosphoriques. Sauf
De plus, le phosphore forme l'acide phosphoreux H3PO3, l'acide phosphorique H4P2O6 et l'acide hypophosphoreux H3PO2, ainsi que des peracides : l'acide perphosphorique H4P2O8 et l'acide monoperphosphorique H3PO5. Les sels d'acides phosphoriques (phosphates) sont largement utilisés. , dans une moindre mesure - phosphites et hypophosphites. Le phosphore se combine directement avec tous les halogènes pour libérer une grande quantité
quantité de chaleur et formation de trihalogénures (PX3, où X est un halogène), de pentahalogénures (PX5) et d'oxyhalogénures (par exemple, POX3) . Lorsque le phosphore fusionne avec le soufre en dessous de 100 °C, des solutions solides à base de phosphore et de soufre se forment, et au-dessus de 100 °C, une réaction exothermique se produit pour former des sulfures cristallins P4S3, P4S5, P4S7, P4S10, dont seul P4S5 se décompose en P4S3 lorsqu'il est chauffé. au-dessus de 200 °C et P4S7, et le reste fond sans décomposition. Les oxysulfures de phosphore suivants sont connus : P2O3S2, P2O3S3, P4O4S3, P6O10S5 et P4O4S3. Le phosphore, comparé à l'azote, est moins capable de former des composés avec l'hydrogène. L’hydrogène phosphine phosphorée PH3 et la diphosphine P2H4 ne peuvent être obtenues qu’indirectement. Parmi les composés phosphore-azote, les nitrures PN, P2N3, P3N5 sont connus - des substances solides et chimiquement stables obtenues en faisant passer de l'azote avec de la vapeur de phosphore à travers un arc électrique ; Halogénures de phosphonitrile polymères - (PNX2)n (par exemple, chlorure de polyphosphonitrile) , obtenu en faisant réagir des pentahalogénures avec de l'ammoniac dans diverses conditions ; les amidoimidophosphates sont des composés, généralement polymères, contenant des liaisons P-NH-P ainsi que des liaisons P-O-P. À des températures supérieures à 2 000 °C, le phosphore réagit avec le carbone pour former
Le carbure PC3 est une substance insoluble dans les solvants ordinaires et n'interagit ni avec les acides ni avec les alcalis. Lorsqu'il est chauffé avec des métaux, le phosphore forme des phosphures. Le phosphore forme de nombreux composés organophosphorés. Reçu. la production de phosphore élémentaire est réalisée
réduction électrothermique de phosphates naturels (apatites ou phosphorites) à 1400-1600 °C avec du coke en présence de silice (sable de quartz) :
2Ca3(PO4)2 + 10C + n SiO2 = P4 + 10CO + 6CaO× n SiO2 Le minerai contenant du phosphore pré-broyé et enrichi est mélangé à
rapports spécifiés avec de la silice et du coke et chargé dans un four électrique. La silice est nécessaire pour réduire la température de la réaction, ainsi que pour augmenter sa vitesse en liant l'oxyde de calcium libéré lors du processus de réduction en silicate de calcium, qui est continuellement éliminé sous forme de scories fondues. Les silicates et oxydes d'aluminium, de magnésium, de fer et d'autres impuretés, ainsi que le ferrophosphore (Fe2P, FeP, Fe3P), formés par l'interaction d'une partie du fer réduit avec le phosphore, passent également dans les scories. Le ferrophosphore, ainsi que de petites quantités de phosphures de manganèse et d'autres métaux dissous, sont retirés du four électrique au fur et à mesure qu'ils s'accumulent pour être utilisés ultérieurement dans la production d'aciers spéciaux. Les vapeurs de phosphore quittent le four électrique avec les sous-produits gazeux.
produits et impuretés volatiles (CO, SiF4, PH3, vapeur d'eau, produits de pyrolyse d'impuretés de charge organique, etc.) à une température de 250-350 °C. Après dépoussiérage, les gaz contenant du phosphore sont envoyés vers des unités de condensation, dans lesquelles le phosphore blanc technique liquide est collecté sous l'eau à une température d'au moins 50 °C. Des méthodes sont en cours de développement pour produire du phosphore à l'aide de gaz
agents réducteurs, réacteurs à plasma afin d'intensifier la production en augmentant les températures jusqu'à 2 500-3 000 °C, c'est-à-dire supérieures aux températures de dissociation des phosphates naturels et des gaz réducteurs (par exemple le méthane) utilisés comme gaz de transport dans le plasma à basse température.
Application. La majeure partie du phosphore produit est transformée en acide phosphorique et en engrais phosphorés et sels techniques (phosphates) obtenus sur cette base. . Le phosphore blanc est utilisé dans les obus incendiaires et fumigènes, les bombes ; rouge
phosphore - dans la production d'allumettes. Le phosphore est utilisé dans la production d’alliages de métaux non ferreux comme agent désoxydant. L'introduction de jusqu'à 1 % de phosphore augmente la résistance thermique des alliages tels que le féchral et le chromal. Le phosphore est inclus dans certains bronzes, car augmente leur fluidité et leur résistance à l'abrasion. Les phosphures de métaux, ainsi que certains non-métaux (B, Si, As, etc.) sont utilisés dans la production et le dopage de matériaux semi-conducteurs. Le phosphore est partiellement utilisé pour produire des chlorures et des sulfures, qui servent de matières premières pour la production de plastifiants contenant du phosphore (par exemple, le phosphate de tricrésyle, le phosphate de tributyle, etc.), de médicaments, de pesticides organophosphorés et sont également utilisés comme additifs dans les lubrifiants. et les carburants. Précautions de sécurité. Le phosphore blanc et ses composés sont hautement toxiques. Travailler avec
le phosphore nécessite une étanchéité minutieuse de l'équipement ; stocker le phosphore blanc. doit être sous l’eau ou dans un récipient métallique hermétiquement fermé. Lorsque vous travaillez avec du phosphore, les règles de sécurité doivent être strictement respectées. Phosphore dans le corps. Le phosphore est l'un des éléments biogènes les plus importants,
nécessaire à la vie de tous les organismes. Il est présent dans les cellules vivantes sous forme d'acides ortho- et pyrophosphoriques et de leurs dérivés, et fait également partie des nucléotides, des acides nucléiques, des phosphoprotéines, des phospholipides, des esters phosphorés de glucides, de nombreux coenzymes et autres composés organiques. En raison des particularités de leur structure chimique, les atomes de phosphore, comme les atomes de soufre, sont capables de former des liaisons riches en énergie dans des composés à haute énergie ; acide adénosine triphosphorique (ATP), créatine phosphate, etc. Au cours du processus d'évolution biologique, ce sont les composés du phosphore qui sont devenus les principaux stockeurs universels d'informations génétiques et de vecteurs d'énergie dans tous les systèmes vivants. Dr. Le rôle important des composés du phosphore dans l'organisme réside dans le fait que l'ajout enzymatique d'un résidu phosphoryle () à divers composés organiques (phosphorylation) sert de « passage » à leur participation au métabolisme, et, à l'inverse, au clivage d'un le résidu phosphoryle (déphosphorylation) exclut ces composés de l'échange actif. Enzymes du métabolisme du phosphore - kinases, phosphorylases et phosphatases. Le foie joue le rôle principal dans la transformation des composés du phosphore dans l’organisme des animaux et des humains. Le métabolisme des composés du phosphore est régulé par les hormones et la vitamine D. Teneur en phosphore (en mg pour 100 g matière sèche) dans les tissus végétaux - 230-350,
animaux marins - 400-1800, animaux terrestres - 1700-4400, bactéries - environ 3000 ; dans le corps humain, il y a surtout beaucoup de phosphore dans le tissu osseux (un peu plus de 5 000), dans le tissu cérébral (environ 4 000) et dans les muscles (220-270). Les besoins humains quotidiens en phosphore sont de 1 à 1,2 g(chez les enfants, il est plus élevé que chez les adultes). Parmi les produits alimentaires, les plus riches en phosphore sont le fromage, la viande, les œufs, les légumineuses (pois, haricots, etc.). L'équilibre du phosphore dans l'organisme dépend de l'état général du métabolisme. La violation du métabolisme du phosphore entraîne de profonds changements biochimiques, principalement dans le métabolisme énergétique. Avec un manque de phosphore dans le corps, les animaux et les humains développent de l'ostéoporose et d'autres maladies osseuses, et les plantes développent une famine en phosphore. . La source de phosphore dans la nature vivante réside dans ses composés inorganiques contenus dans le sol et dissous dans l'eau. Le phosphore est extrait du sol par les plantes sous forme de phosphates solubles. Les animaux consomment généralement suffisamment de phosphore dans leur alimentation. Après la mort des organismes, le phosphore pénètre à nouveau dans le sol et les sédiments du fond, participant ainsi. dans le cycle des substances. Le rôle important du phosphore dans la régulation des processus métaboliques détermine la grande sensibilité de nombreux systèmes enzymatiques de cellules vivantes à l'action des composés organophosphorés. Cette circonstance est utilisée en médecine dans le développement de médicaments, en agriculture dans la production de phosphore.
engrais, ainsi que dans la création d'insecticides efficaces. De nombreux composés du phosphore sont extrêmement toxiques et certains composés organophosphorés peuvent être classés comme agents de guerre chimique (sarin, soman, tabun). L'isotope radioactif du phosphore 32P est largement utilisé en biologie et en médecine comme indicateur dans l'étude de tous les types de métabolisme et d'énergie dans les organismes vivants. . Médicaments contenant du phosphore (acide adénosine triphosphorique,
phytine, glycérophosphate de calcium, phosphrène, etc.), affectent principalement les processus du métabolisme tissulaire et sont utilisés pour les maladies des muscles, du système nerveux, la tuberculose, la perte de nutrition, l'anémie, etc. Les isotopes radioactifs du phosphore sont utilisés comme indicateurs isotopiques pour étudier le métabolisme, le diagnostic des maladies, ainsi que la radiothérapie des tumeurs .
Arsenic(lat. Arsenicum), As, élément chimique du groupe V du système périodique de Mendeleev, numéro atomique 33, masse atomique 74,9216 ; cristaux gris acier. L'élément est constitué d'un isotope stable 75As. Référence historique. Composés naturels d'arsenic avec du soufre (orpiment As2S3,
realgar As4S4) étaient connus des peuples du monde antique, qui utilisaient ces minéraux comme médicaments et peintures. Le produit de la combustion des sulfures d'arsenic était également connu - l'oxyde d'arsenic (III) As2O3 («arsenic blanc»). Le nom d'arsenikón se retrouve déjà chez Aristote ; il est dérivé du grec. ársen - fort, courageux et servait à désigner les composés d'arsenic (en fonction de leur fort effet sur le corps). On pense que le nom russe viendrait de « souris » (d’après l’utilisation de préparations à base d’arsenic pour exterminer les souris et les rats). La production d'arsenic à l'état libre est attribuée à Albert le Grand (vers 1250). En 1789 A. Lavoisier inclut l'arsenic dans la liste des éléments chimiques. Répartition dans la nature. Teneur moyenne en arsenic dans la croûte terrestre (Clark)
1,7·10-4% (en masse), en telles quantités, il est présent dans la plupart des roches ignées. Étant donné que les composés d'arsenic sont volatils à haute température, l'élément ne s'accumule pas lors des processus magmatiques ; il se concentre en précipitant à partir des eaux chaudes et profondes (avec S, Se, Sb, Fe, Co, Ni, Cu et d'autres éléments). Lors des éruptions volcaniques, l'arsenic sous forme de composés volatils pénètre dans l'atmosphère. L’arsenic étant multivalent, sa migration est fortement influencée par l’environnement rédox. Dans les conditions oxydantes de la surface terrestre, des arséniates (As5+) et des arsénites (As3+) se forment. Ce sont des minéraux rares que l’on trouve uniquement dans les zones de gisements d’arsenic. L’arsenic natif et les minéraux As2+ sont encore moins courants. Parmi les nombreux minéraux d'arsenic (environ 180), seul l'arsénopyrite FeAsS revêt une importance industrielle primordiale. De petites quantités d'arsenic sont essentielles à la vie. Cependant, dans les zones de gisement
l'arsenic et l'activité des jeunes volcans, les sols contiennent à certains endroits jusqu'à 1% d'arsenic, qui est associé aux maladies du bétail et à la mort de la végétation. L'accumulation d'arsenic est particulièrement typique des paysages de steppes et de déserts, dans les sols desquels l'arsenic est inactif. Dans les climats humides, l’arsenic est facilement éliminé du sol. Dans la matière vivante, il y a en moyenne 3,10-5% d'arsenic, dans les rivières 3,10-7%. L'arsenic apporté
rivières dans l'océan, il se stabilise relativement rapidement. Dans l'eau de mer, il n'y a que 1,10 à 7 % d'arsenic, mais dans les argiles et les schistes, il est de 6,6,10 à 4 %. Les minerais de fer sédimentaires et les nodules de ferromanganèse sont souvent enrichis en arsenic.
Proprietes physiques et chimiques. L'arsenic présente plusieurs modifications allotropiques. Dans des conditions normales, le plus stable est ce qu'on appelle l'arsenic métallique, ou gris (a-As) - une masse cristalline cassante gris acier ; lorsqu'il est fraîchement fracturé, il a un éclat métallique ; à l'air, il s'estompe rapidement car il est recouvert d'une fine pellicule d'As2O3. Le réseau cristallin de l'arsenic gris est rhomboédrique ( UN- 4,123 Å, angle a – 54о10", X- 0,226), en couches. Densité 5,72 g/cm3(à 20°C), résistivité électrique 35·10-8 ohm× m, ou 35·10-6 ohm× cm, coefficient de température de résistance électrique 3,9·10-3 (0®-100 оC), dureté Brinell 1470 Mn/m2, ou 147 kgf/mm2(3-4 selon Mohs) ; L'arsenic est diamagnétique. Sous pression atmosphérique, l'arsenic se sublime à 615 °C sans fondre, puisque le point triple (voir diagramme de phases) a-As se situe à 816 °C et à une pression de 36 à. La vapeur d'arsenic est constituée de molécules As4 jusqu'à 800°C, au-dessus de 1700°C - uniquement As2. Lorsque la vapeur d'arsenic se condense sur une surface refroidie par l'air liquide, de l'arsenic jaune se forme - des cristaux transparents et mous comme de la cire, d'une densité de 1,97. g/cm3, dont les propriétés sont similaires à celles du phosphore blanc. Lorsqu'il est exposé à la lumière ou à une faible chaleur, il se transforme en arsenic gris. Des modifications vitreuses-amorphes sont également connues : l'arsenic noir et l'arsenic brun, qui, lorsqu'ils sont chauffés au-dessus de 270 ? C, se transforment en M gris. Configuration des électrons externes de l'atome d'arsenic 3 d 104s 24p 3. Arsenic dans les composés
a les états d'oxydation + 5, + 3 et - 3. L'arsenic gris est beaucoup moins actif chimiquement que le phosphore. Lorsqu’il est chauffé dans l’air à plus de 400°C, l’arsenic brûle, formant As2O3. L'arsenic se combine directement avec les halogènes ; dans des conditions normales, AsF5 est un gaz ; AsF3, AsCl3, AsBr3 - liquides incolores et hautement volatils ; AsI3 et As2l4 sont des cristaux rouges. Lorsque l'arsenic et le soufre sont chauffés, on obtient des sulfures : As4S4 rouge orangé et As2S3 jaune citron. Le sulfure jaune pâle As2S5 est précipité par passage de H2S dans une solution glacée d'acide arsénique (ou de ses sels) dans de l'acide chlorhydrique fumant : 2H3AsO4 + 5H2S = As2S5 + 8H2O ; À environ 500°C, il se décompose en As2S3 et en soufre. Tous les sulfures de M. sont insolubles dans l’eau et les acides dilués. Des agents oxydants forts (mélanges de HNO3 + HCl, HCl + KClO3) les transforment en un mélange de H3AsO4 et H2SO4. Le sulfure As2S3 se dissout facilement dans les sulfures et polysulfures d'ammonium et de métaux alcalins, formant des sels d'acides - thioarsenic H3AsS3 et thioarsenic H3AsS4. Avec l'oxygène, l'arsenic produit des oxydes : oxyde M. (III) As2O3 - anhydride d'arsenic et oxyde M. (V) As2O5 - anhydride d'arsenic. Le premier d'entre eux est formé par l'action de l'oxygène sur le métal ou ses sulfures, par exemple 2As2S3 + 9O2 - 2As2O3 + 6SO2. Les vapeurs d'As2O3 se condensent en une masse vitreuse incolore, qui devient opaque avec le temps en raison de la formation de petits cristaux cubiques, densité 3,865. g/cm3. La densité de vapeur correspond à la formule As4O6 : au-dessus de 1800°C la vapeur est constituée d'As2O3. À 100 g l'eau se dissout 2.1 g As2O3 (à 25°C). M. oxyde (III) est un composé amphotère avec une prédominance de propriétés acides. On connaît des sels (arsénites) correspondant aux acides orthoarsenic H3AsO3 et métaarsenic HAsO2 ; les acides eux-mêmes n'ont pas été obtenus. Seuls les arsénites de métaux alcalins et d’ammonium sont solubles dans l’eau. L'As2O3 et les arsénites sont généralement des agents réducteurs (par exemple, As2O3 + 2I2 + 5H2O - 4HI + 2H3AsO4), mais peuvent également être des agents oxydants (par exemple, As2O3 + 3C - 2As + 3CO). L'oxyde d'arsenic(V) est préparé en chauffant l'acide arsénique H3AsO4 (environ
200°C). Il est incolore et se décompose vers 500°C en As2O3 et O2. L'acide arsénique est préparé par action de HNO3 concentré sur As ou As2O3. Les sels d'acide arsénique (arséniates) sont insolubles dans l'eau, à l'exception des sels de métaux alcalins et d'ammonium. On connaît des sels qui correspondent aux acides orthoarsenic H3AsO4, métaarsenic HAsO3 et pyroarsenic H4As2O7 ; les deux derniers acides n'ont pas été obtenus à l'état libre. Lorsqu’il est allié à des métaux, l’arsenic forme principalement des composés (arséniures).
Réception et demande. L'arsenic est produit industriellement en chauffant des pyrites d'arsenic :
FeAsS - FeS + As ou (moins souvent) réduction d'As2O3 avec du charbon. Les deux processus sont effectués dans des cornues de
argile réfractaire reliée à un récepteur de condensation des vapeurs d'arsenic. L'anhydride d'arsenic est obtenu par grillage oxydatif de minerais d'arsenic ou comme sous-produit du grillage de minerais polymétalliques, qui contiennent presque toujours de l'arsenic. Lors du grillage oxydatif, des vapeurs d'As2O3 se forment et se condensent dans les chambres de collecte. L'As2O3 brut est purifié par sublimation à 500-600°C. L'As2O3 purifié est utilisé pour la production d'arsenic et de ses préparations.
De petits ajouts d'arsenic (0,2 à 1,0 % en poids) sont introduits dans le plomb utilisé pour la production de grenailles d'armes à feu (l'arsenic augmente la tension superficielle du plomb fondu, grâce à quoi la grenaille acquiert une forme proche de sphérique ; l'arsenic augmente légèrement la dureté de plomb). En remplacement partiel de l'antimoine, l'arsenic est inclus dans certains alliages de régule et d'impression. L'arsenic pur n'est pas toxique, mais tous ses composés, solubles dans l'eau ou capables de
passer en solution sous l'influence du suc gastrique, extrêmement toxique ; L'hydrogène arsenic est particulièrement dangereux. Parmi les composés d'arsenic utilisés dans la production, l'anhydride d'arsenic est le plus toxique. Presque tous les minerais sulfurés de métaux non ferreux, ainsi que la pyrite de fer (soufre), contiennent un mélange d'arsenic. Par conséquent, lors de leur grillage oxydatif, avec le dioxyde de soufre SO2, il se forme toujours As2O3 ; La majeure partie se condense dans les canaux de fumée, mais en l'absence ou en cas de faible efficacité des installations de traitement, les gaz d'échappement des fours de grillage du minerai emportent des quantités notables d'As2O3. L'arsenic pur, bien que non toxique, est toujours recouvert d'une couche d'As2O3 toxique lorsqu'il est stocké dans l'air. En l'absence d'une ventilation adéquate, la gravure des métaux (fer, zinc) avec des acides sulfuriques ou chlorhydriques industriels contenant de l'arsenic est extrêmement dangereuse, car elle produit de l'hydrogène arsenic. Arsenic dans le corps. En tant qu’oligoélément, l’arsenic est omniprésent
dans la nature vivante. La teneur moyenne en arsenic dans les sols est de 4,10 à 4 %, dans les cendres végétales de 3,10 à 5 %. La teneur en arsenic des organismes marins est plus élevée que celle des organismes terrestres (chez les poissons 0,6-4,7 mg en 1 kg la matière brute s'accumule dans le foie). La teneur moyenne en arsenic dans le corps humain est de 0,08 à 0,2 mg/kg. Dans le sang, l'arsenic est concentré dans les globules rouges, où il se lie à la molécule d'hémoglobine (et la fraction globine en contient deux fois plus que l'hème). La plus grande quantité (pour 1 g tissu) se trouve dans les reins et le foie. Une grande quantité d'arsenic se trouve dans les poumons et la rate, la peau et les cheveux ; relativement peu - dans le liquide céphalo-rachidien, le cerveau (principalement l'hypophyse), les gonades, etc. Dans les tissus, l'arsenic se trouve dans la fraction protéique principale, beaucoup moins dans la fraction acidosoluble, et seule une petite partie de celui-ci se trouve dans la fraction lipidique. L'arsenic est impliqué dans les réactions redox : dégradation oxydative des glucides complexes, fermentation, glycolyse, etc. Les composés de l'arsenic sont utilisés en biochimie comme inhibiteurs d'enzymes spécifiques pour étudier les réactions métaboliques. L'arsenic en médecine. Composés organiques de l'arsenic (aminarsone, miarsenol,
norasénal, osarsol) sont principalement utilisés pour le traitement de la syphilis et des maladies à protozoaires. Les préparations inorganiques d'arsenic - arsénite de sodium (acide arsénique de sodium), arsénite de potassium (acide arséniique de potassium), anhydride d'arsenic As2O3, sont prescrites comme agents fortifiants et toniques généraux. Lorsqu'elles sont appliquées localement, les préparations d'arsenic inorganique peuvent provoquer un effet nécrosant sans irritation préalable, ce qui rend ce processus presque indolore ; cette propriété, la plus prononcée dans As2O3, est utilisée en dentisterie pour
destruction de la pulpe dentaire. Des préparations d'arsenic inorganique sont également utilisées pour traiter le psoriasis. Isotopes radioactifs obtenus artificiellement M. 74As (T1/2 - 17,5 jours) et 76As (T1/2
26,8 h) sont utilisés à des fins diagnostiques et thérapeutiques. Avec leur aide, la localisation des tumeurs cérébrales est clarifiée et le degré de radicalité de leur ablation est déterminé. L'arsenic radioactif est parfois utilisé pour les maladies du sang, etc. Selon les recommandations de la Commission internationale de radioprotection,
teneur maximale admissible de 76As dans le corps 11 McCurie. Selon les normes sanitaires adoptées en URSS, les concentrations maximales admissibles de 76As dans l'eau et les réservoirs ouverts sont de 1,10-7. curie/l, dans l'air des locaux de travail 5·10-11 curie/l. Toutes les préparations à base d'arsenic sont très toxiques. En cas d'intoxication aiguë, des douleurs abdominales sévères, de la diarrhée et des lésions rénales sont observées ; Des effondrements et des convulsions sont possibles. Dans les intoxications chroniques, les plus fréquents sont les troubles gastro-intestinaux, le catarrhe des muqueuses des voies respiratoires (pharyngite, laryngite, bronchite), les lésions cutanées (exanthème, mélanose, hyperkératose) et les troubles de la sensibilité ; le développement d'une anémie aplasique est possible. Dans le traitement des intoxications par les préparations à base d'arsenic, l'unithiol est de la plus haute importance. Les mesures de prévention des intoxications industrielles devraient viser à
principalement sur la mécanisation, l'étanchéité et le dépoussiérage du processus technologique, en créant une ventilation efficace et en fournissant aux travailleurs un équipement de protection individuelle contre l'exposition à la poussière. Des examens médicaux réguliers des travailleurs sont nécessaires. Des examens médicaux préliminaires sont effectués à l'embauche et pour les salariés - une fois tous les six mois.
ANTIMOINE(lat. Stibium), Sb, élément chimique du groupe V du système périodique de Mendeleïev ; numéro atomique 51, masse atomique 121,75 ; Le métal est blanc argenté avec une teinte bleutée. Deux isotopes stables sont connus dans la nature : 121Sb (57,25 %) et 123Sb (42,75 %). Parmi les isotopes radioactifs obtenus artificiellement, le plus important est le 122Sb ( T 1/2 - 2,8 Cym), 124Sb ( T 1/2 - 60,2 Cym) et 125Sb ( T 1/2 - 2 ans).
Référence historique. L'antimoine est connu depuis l'Antiquité. Dans les pays de l’Est, il était utilisé vers 3000 avant JC. e. pour fabriquer des vases. Dans l'Egypte ancienne déjà au 19ème siècle. avant JC e. De la poudre pailletée d'antimoine (Sb2S3 naturel) appelée mesten ou tige était utilisée pour noircir les sourcils. Dans la Grèce antique, on l'appelait stími et stíbi, d'où le latin stibium. Vers 12-14 siècles. n. e. le nom d'antimoine est apparu. En 1789, A. Lavoisier inclut l'antimoine dans la liste des éléments chimiques appelés antimoine (antimoine anglais moderne, antimonio espagnol et italien, antimon allemand). « antimoine » russe vient du turc sürme ; il désignait la poudre de paillettes de plomb PbS, qui était également utilisée pour noircir les sourcils (selon d'autres sources, « antimoine » - du persan surme - métal). Une description détaillée des propriétés et des méthodes de production de l'antimoine et de ses composés a été donnée pour la première fois par l'alchimiste Vasily Valentin (Allemagne) en 1604. Répartition dans la nature. Teneur moyenne en antimoine dans la croûte terrestre (clarke) 5 ×10-5
% par poids. L'antimoine est dispersé dans le magma et la biosphère. Issu des eaux souterraines chaudes, il se concentre dans les gisements hydrothermaux. Les gisements d'antimoine eux-mêmes sont connus, ainsi que les gisements d'antimoine-mercure, d'antimoine-plomb, d'or-antimoine et d'antimoine-tungstène. Parmi les 27 minéraux d’antimoine, la stibine (Sb2S3) revêt une importance industrielle majeure. . En raison de son affinité avec le soufre, l'antimoine se trouve souvent comme impureté dans les sulfures d'arsenic, de bismuth, de nickel, de plomb, de mercure, d'argent et d'autres éléments. Proprietes physiques et chimiques. L'antimoine est connu sous forme cristalline et trois
formes amorphes (explosive, noire et jaune). Antimoine explosif (densité 5,64-5,97 g/cm 3) explose à tout contact : formé lors de l'électrolyse d'une solution
SbCl3; noir (densité 5,3 g/cm 3) - avec refroidissement rapide des vapeurs d'antimoine ; jaune - lorsque l'oxygène passe dans le SbH3 liquéfié. L'antimoine jaune et noir est instable ; à basse température, ils se transforment en antimoine ordinaire. L'antimoine cristallin le plus stable , cristallise dans le système trigonal, UN - 4,5064 ; densité 6,61-6,73 g/cm 3(liquide - 6,55 g/cm 3); t point de fusion 630,5 °C ; t kip1635-1645 °C ; capacité thermique spécifique à 20-100 °C 0,210 kJ/(kg ×À ) ; conductivité thermique à 20 °C 17,6 F/M× K. Coefficient de température de dilatation linéaire pour le C polycristallin. 11,5 × 10-6 à 0-100 °C ; pour monocristal a1 - 8,1 × 10-a2 - 19,5×10-6 à 0-400 °C, résistivité électrique (20 °C) (43,045×10-6 ohm× cm). L'antimoine est diamagnétique, la susceptibilité magnétique spécifique est de -0,66 ×10-6. Contrairement à la plupart des métaux, l'antimoine est fragile, se divise facilement le long des plans de clivage, se réduit en poudre et ne peut pas être forgé (il est parfois classé comme semi-métal). . Les propriétés mécaniques dépendent de la pureté du métal. Dureté Brinell pour métal moulé 325-340 Mn/m 2 (32,5-34,0 kgf/mm 2); module élastique 285-300 ; résistance à la traction 86,0 Mn/m 2 (8,6 kgf/mm 2). Configuration des électrons externes de l'atome Sb5s25r3. Dans les composés, il présente principalement des états d'oxydation +5, +3 et -3. Chimiquement, l'antimoine est inactif. Ne s'oxyde pas à l'air jusqu'à ce que
température de fusion. L'antimoine ne réagit pas avec l'azote et l'hydrogène. Le carbone se dissout légèrement dans l'antimoine fondu. Le métal interagit activement avec le chlore et d'autres halogènes, formant des halogénures d'antimoine. Réagit avec l'oxygène à des températures supérieures à 630 °C pour former du Sb2O3 . Lorsqu'ils sont fusionnés avec du soufre, des sulfures d'antimoine sont obtenus et interagissent également avec le phosphore et l'arsenic. L'antimoine résiste à l'eau et aux acides dilués. Les acides chlorhydrique et sulfurique concentrés dissolvent lentement l'antimoine pour former du chlorure SbCl3 et du sulfate Sb2 (SO4)3 ; l'acide nitrique concentré oxyde l'antimoine en un oxyde supérieur, qui se forme sous la forme d'un composé hydraté xSb2O5 × yH2O. Les sels peu solubles de l'acide d'antimoine - les antimoniates (MeSbO3 × 3H2O, où Me est Na, K) et les sels d'acide métaantimoine non isolé - les métaantimonites (MeSbO2 × 3H2O), qui ont des propriétés réductrices, présentent un intérêt pratique. S. se combine avec les métaux pour former des antimoniures. Reçu. L'antimoine est obtenu par des méthodes pyrométallurgiques et hydrométallurgiques
traitement de concentrés ou de minerais contenant 20 à 60 % de Sb. Les méthodes pyrométallurgiques comprennent la fusion par précipitation et par réduction. Les matières premières pour la fusion par précipitation sont des concentrés de sulfures ; le procédé est basé sur le déplacement de l'antimoine de son sulfure par le fer : Sb2S3 + 3Fe Û 2Sb + 3FeS. Le fer est introduit dans la charge sous forme de ferraille. La fusion est réalisée dans des fours à réverbère ou à tambour rotatif court à 1 300-1 400 °C. La récupération de l'antimoine en métal brut est supérieure à 90 %. La fusion réductrice de l'antimoine est basée sur la réduction de ses oxydes en métal avec du charbon de bois ou de la poussière de charbon et des scories de stériles. La fusion par réduction est précédée d'une torréfaction oxydative à 550 °C avec un excès d'air. Les cendres contiennent du tétroxyde d'antimoine non volatil. Les fours électriques peuvent être utilisés à la fois pour les matières fondues par précipitation et par réduction. La méthode hydrométallurgique de production d'antimoine comprend deux étapes : le traitement de la matière première avec une solution de sulfure alcalin, le transfert de l'antimoine en solution sous forme de sels d'acides d'antimoine et de sulfosels et la séparation de l'antimoine. électrolyse. L'antimoine brut, selon la composition de la matière première et son mode de production, contient de 1,5 à 15 % d'impuretés : Fe, As, S, etc. Pour obtenir de l'antimoine pur, un raffinage pyrométallurgique ou électrolytique est utilisé. Lors du raffinage pyrométallurgique, les impuretés de fer et de cuivre sont éliminées sous forme de composés soufrés en introduisant de l'antimonite (crudum) - Sb2S3 - dans la masse fondue d'antimoine, après quoi l'arsenic (sous forme d'arséniate de sodium) et le soufre sont éliminés en soufflant de l'air sous le scories de soude. Lors du raffinage électrolytique avec une anode soluble, l'antimoine brut est purifié du fer, du cuivre et des autres métaux restant dans
électrolyte (Cu, Ag, Au restent dans les boues). L'électrolyte est une solution composée de SbF3, H2SO4 et HF. La teneur en impuretés de l'antimoine raffiné ne dépasse pas 0,5 à 0,8 %. Pour obtenir de l'antimoine de haute pureté, la fusion de zone est utilisée dans une atmosphère de gaz inerte ou l'antimoine est obtenu à partir de composés pré-purifiés - trioxyde ou trichlorure. Application. L'antimoine est utilisé principalement sous forme d'alliages de plomb et d'étain
pour plaques de batterie, gaines de câbles, roulements (babbitt) , alliages utilisés dans l'imprimerie (hart) , etc. Ces alliages ont une dureté, une résistance à l'usure et une résistance à la corrosion accrues. Dans les lampes fluorescentes, Sb est activé avec de l'halophosphate de calcium. L'antimoine est inclus dans les matériaux semi-conducteurs comme dopant au germanium et au silicium, ainsi que dans les antimoniures (par exemple InSb). L'isotope radioactif 12Sb est utilisé dans les sources de rayonnement g et de neutrons. Antimoine dans le corps. Teneur en antimoine (pour 100 g matière sèche) est en
plantes 0,006 mg, chez les animaux marins 0,02 mg, chez les animaux terrestres 0,0006 mg. L'antimoine pénètre dans le corps des animaux et des humains par le système respiratoire ou le tractus gastro-intestinal. Il est excrété principalement dans les selles et en petites quantités dans l'urine. Le rôle biologique de l'antimoine est inconnu. Il est sélectivement concentré dans la glande thyroïde, le foie et la rate. Dans les érythrocytes, l'antimoine s'accumule principalement à l'état d'oxydation + 3, dans le plasma sanguin - à l'état d'oxydation + 5. Concentration maximale admissible C. 10-5 - 10-7 g par 100 g linge sec. À des concentrations plus élevées, cet élément inactive un certain nombre d'enzymes du métabolisme des lipides, des glucides et des protéines (peut-être en raison du blocage des groupes sulfhydryle). . Dans la pratique médicale, les préparations d'antimoine (solyusurmine, etc.) sont utilisées dans
principalement pour le traitement de la leishmaniose et de certaines helminthiases (par exemple la schistosomiase). L'antimoine et ses composés sont toxiques. L'empoisonnement est possible lors de la fusion du concentré
minerais d'antimoine et dans la production d'alliages d'antimoine. En cas d'intoxication aiguë - irritation des muqueuses des voies respiratoires supérieures, des yeux et de la peau. Des dermatites, conjonctivites, etc. peuvent se développer. Traitement : antidotes (unithiol), diurétiques et diaphorétiques, etc. Prévention : mécanisation de la production. processus, ventilation efficace, etc.
BISMUTH(lat. Bismuthum), Bi, élément chimique du groupe périodique V
le système de Mendeleïev ; numéro atomique 83, masse atomique 208,980 ; métal gris argenté avec une teinte rosée. Le bismuth naturel est constitué d'un isotope stable, le 209Bi. V. était connu aux XVe et XVIe siècles, mais il a longtemps été considéré comme un type d'étain,
plomb ou antimoine. Le bismuth a été reconnu comme métal indépendant au milieu du XVIIIe siècle. Le chimiste français A. Lavoisier l'a inclus dans la liste des corps simples. L’origine du nom « bismuth » n’est pas établie. La teneur en bismuth de la croûte terrestre est de 2,10 à 5 % en masse. Le bismuth est présent naturellement dans
sous forme de nombreux minéraux dont les plus importants sont le bismuth lustre Bi2S3, le bismuth natif Bi, la bismite Bi2O3, etc. En plus grande quantité, mais en faibles concentrations, le bismuth se retrouve comme impureté isomorphe dans le plomb-zinc, le cuivre, le molybdène -les minerais de cobalt et d'étain-tungstène. Environ 90 % de la consommation mondiale est couverte par la production associée de bismuth lors du traitement des minerais polymétalliques. Proprietes physiques et chimiques. Le bismuth possède un réseau rhomboédrique avec
période un- 4,7457 A et angle α-57?14 " 13"" . Densité 9,80 g/cm 3; t température ambiante : 271,3 °C ; t température ambiante : 1560 °C. Capacité thermique spécifique (20 °C) 123,5 J/kg K (0,0294 calories/g·AVEC); coefficient thermique
expansion linéaire à température ambiante 13,3·10-6 ; conductivité thermique (20°C) 8,37 W/(m·À) ; résistivité électrique (20°C) 106,8 10-8 ohm· m(106,8·10-6 ohm· cm). Le bismuth est le métal le plus diamagnétique. La susceptibilité magnétique spécifique est de -1,35·10-6. Sous l'influence d'un champ magnétique, la résistance électrique du bismuth augmente davantage que celle des autres métaux, ce qui permet de mesurer l'induction de champs magnétiques puissants. La section efficace de capture des neutrons thermiques du bismuth est petite (34 10-31 m 2 ou 0,034 grange). À température ambiante, le bismuth est fragile, se divise facilement le long des plans de clivage et est réduit en poudre dans un mortier de porcelaine. Forgeage à une température de 120-150°C ; par pressage à chaud (à 240-250°C), il peut être utilisé pour produire du fil d'un diamètre allant jusqu'à 0,1 mm, ainsi que des plaques d'une épaisseur de 0,2 à 0,3 mm. Dureté Brinell 93 Mn/m 2 (9,3 kgf/mm 2), selon Mohs 2.5. Lors de la fusion, V. diminue de volume de 3,27 %. Le bismuth est stable dans l'air sec, mais dans l'air humide sa surface
oxydation. Lorsqu'il est chauffé au-dessus de 1 000 °C, il brûle avec une flamme bleuâtre pour former de l'oxyde Bi2O3. . Dans la série de tension, le bismuth se situe entre l'hydrogène et le cuivre, il ne se dissout donc pas dans les acides sulfurique et chlorhydrique dilués ; la dissolution dans les acides sulfurique et nitrique concentrés se produit avec la libération de SO2 et d'oxydes d'azote correspondants. Le bismuth présente des valences de 2, 3 et 5. Composés du bismuth de valences inférieures
ont un caractère basique, les plus élevés sont acides. Parmi les composés oxygénés du monde, le plus important est le trioxyde Bi2O3, qui change de couleur jaune en rouge-brun lorsqu'il est chauffé. Bi2O3 est utilisé pour obtenir des sels de bismuth. Dans les solutions diluées, les sels de bismuth s'hydrolysent. Le chlorure de BiCl3 est hydrolysé avec la précipitation de l'oxychlorure BiOCl, du nitrate de Bi (NO3)3 - avec la précipitation du sel principal BiONO3·BiOOH. La capacité des sels de V. à s'hydrolyser est utilisée pour sa purification. Les composés du bismuth 5-valent sont difficiles à obtenir ; ce sont de puissants agents oxydants. Le sel KBiO3 (correspondant à l'anhydride Bi2O5) se forme sous forme d'un précipité brun-rouge sur une anode en platine lors de l'électrolyse d'une solution bouillante d'un mélange de KOH, KCl et d'une suspension de Bi2O3. Le bismuth se combine facilement avec les halogènes et le soufre. Lorsque les acides agissent sur un alliage de bismuth et de magnésium, il se forme de la bismuthine (hydrogène du bismuth) BiH3 ; Contrairement à l'arsine AsH3, la bismuthine est un composé instable et n'a pas été obtenue sous sa forme pure (sans excès d'hydrogène). Avec certains métaux (plomb, cadmium, étain), le bismuth forme des eutectiques à bas point de fusion ; avec du sodium, du potassium, du magnésium et du calcium - composés intermétalliques avec un point de fusion nettement supérieur au point de fusion des composants d'origine. Le bismuth n'interagit pas avec l'aluminium, le chrome et le fer fondus. Réception et demande. La majeure partie du bismuth est produite comme sous-produit
raffinage au feu du plomb brut (werkbley). La méthode pyrométallurgique repose sur la capacité du bismuth à former des composés intermétalliques réfractaires avec K, Na, Mg et Ca. Les métaux spécifiés sont ajoutés au plomb fondu et les composés solides résultants avec V. (crasses) sont séparés de la masse fondue. Une quantité importante de bismuth est extraite des boues issues du raffinage électrolytique du plomb dans une solution hydrofluorosilicique, ainsi que des poussières et boues issues de la production de cuivre. Les crasses et boues contenant du bismuth sont fondues sous des scories alcalines. Le métal brut résultant contient des impuretés As, Sb, Cu, Pb, Zn, Se, Te, Ag et quelques autres éléments. La fusion du bismuth à partir de ses propres minerais est réalisée à petite échelle. Les minerais sulfurés sont traités par fusion par précipitation avec de la ferraille. À partir de minerais oxydés, l'or est réduit avec du charbon sous une couche de fondant à faible point de fusion. Pour le nettoyage grossier des eaux brutes, ils sont utilisés en fonction de la composition des impuretés.
diverses méthodes : zigérisation, affinage oxydatif sous flux alcalins, fusion avec du soufre, etc. L'impureté du plomb la plus difficile à séparer est éliminée (jusqu'à 0,01 %) en soufflant du chlore à travers le métal en fusion. Le bismuth commercial contient
99,9 à 99,98 % de métaux communs. V. de haute pureté sont obtenus par recristallisation de zone dans des cuves en quartz sous atmosphère de gaz inerte. Une quantité importante de bismuth est utilisée pour la préparation d'alliages à bas point de fusion contenant du plomb, de l'étain, du cadmium, qui sont utilisés dans les prothèses dentaires, pour la fabrication de clichés à partir de matrices en bois, comme capuchons fondants dans les dispositifs automatiques de lutte contre l'incendie, lors du soudage de capuchons sur des projectiles perforants, etc. Le bismuth fondu peut servir de liquide de refroidissement dans les réacteurs nucléaires. La consommation de bismuth dans les composés avec Te pour
générateurs thermoélectriques. Ces composés, grâce à la combinaison favorable de conductivité thermique, de conductivité électrique et de force thermoélectromotrice, permettent de convertir l'énergie thermique en énergie électrique avec un rendement élevé (~7 %). L'ajout de bismuth aux aciers inoxydables améliore leur usinabilité. Les composés de bismuth sont utilisés dans la fabrication du verre, de la céramique et des produits pharmaceutiques.
industrie. Les sels de bismuth solubles sont toxiques et ont des effets similaires à ceux du mercure.
Littérature
1. Substances nocives dans l'industrie, en général. éd. N. V. Lazareva, 6e éd., partie 2, Leningrad, 1971.
2. Ivanov V.V. Géochimie écologique des éléments. 3. Recherche dans le domaine de la création d'une nouvelle technologie pour la production d'antimoine et de ses
composés, dans la collection : Chimie et technologie de l'antimoine, France, 1965. 4. Brève encyclopédie chimique, vol 5, M., 1967 ; 5. Nekrasov B.V., Fondamentaux de chimie générale, vol. 1, M., 1965 ; 6. Pogodin S.A., Arsenic, dans l'ouvrage : Brève encyclopédie chimique, vol 3, M., 1964 7. Rémy G., Cours de chimie inorganique, trad. de l'allemand, vol. 1, M., 1963, p. 700-712 ; 8. Thomson J.G., Bismuth, trad. [de l'anglais], L., 1932 ; 9. Shiyanov A.G., Production d'antimoine, M., 1961 ; Fondements de la métallurgie, vol. 5, M.,
1968 ; 10. Chimie et technologie de l'azote lié, [M.-L.], 1934 ; KHE, tome 1, M., 1961.
Le sous-groupe de l'azote se compose de cinq éléments : l'azote, le phosphore, l'arsenic, l'antimoine et le bismuth. Ce sont des éléments p du groupe V du système périodique de D.I. Mendeleïev.
Au niveau d'énergie externe, les atomes de ces éléments contiennent cinq électrons, qui ont la configuration ns2np3 et sont répartis comme suit :

Par conséquent, l'état d'oxydation le plus élevé de ces éléments est +5, le plus bas est -3 et +3 est également typique.
La présence de trois électrons non appariés au niveau externe indique que dans un état non excité, les atomes des éléments ont une valence de 3. Le niveau externe de l'atome d'azote se compose de seulement deux sous-niveaux - 2s et 2p. Les atomes des éléments restants de ce sous-groupe ont des cellules vacantes du sous-niveau d aux niveaux d'énergie externes. Par conséquent, l'un des électrons s du niveau externe peut, lors de l'excitation, se déplacer vers le sous-niveau d du même niveau, ce qui conduit à la formation de 5 électrons non appariés.

couche électronique externe de phosphore (atome non excité)

la couche électronique externe d’un atome de phosphore excité.
Ainsi, le phosphore, l'arsenic, l'antimoine et le bismuth dans un état excité ont 5 électrons non appariés, et leur valence dans cet état est de 5.
Dans un atome d'azote, il est impossible d'exciter un électron de cette manière en raison de l'absence de sous-niveau d au deuxième niveau. Par conséquent, l’azote ne peut pas être pentavalent, mais il peut former une quatrième liaison covalente par le mécanisme donneur-accepteur en raison de la paire électronique solitaire 2s2. Un autre processus est également possible pour l’atome d’azote. Lorsqu’un des deux électrons 2s est retiré, l’azote se transforme en un ion tétravalent N+ à charge unique.

De l'azote au bismuth, les rayons atomiques augmentent et les potentiels d'ionisation diminuent. Les propriétés réductrices des atomes neutres augmentent de N à Bi et les propriétés oxydantes s'affaiblissent (voir tableau 21).
Avec l'hydrogène, l'azote, le phosphore et l'arsenic forment des composés polaires RH3, présentant un état d'oxydation négatif de -3. Les molécules RH3 ont une forme pyramidale. Dans ces composés, les liaisons des éléments avec l'hydrogène sont plus fortes que dans les composés correspondants des éléments du sous-groupe oxygène et surtout du sous-groupe halogène. Par conséquent, les composés hydrogènes des éléments du sous-groupe azoté dans les solutions aqueuses ne forment pas d'ions hydrogène.

Avec l'oxygène, les éléments du sous-groupe azote forment des oxydes de formule générale R2O3 et R2O5. Les oxydes correspondent aux acides HRO2 et HRO3 (et aux orthoacides H3RO4, sauf l'azote). Au sein du sous-groupe, la nature des oxydes change comme suit : N2O3 - oxyde acide ; Р4О6 - oxyde faiblement acide ; As2O3 est un oxyde amphotère aux propriétés acides prédominantes ; Sb2O3 est un oxyde amphotère avec une prédominance de propriétés basiques ; Bi2O3 est le principal oxyde. Ainsi, les propriétés acides des oxydes de composition R2O3 et R2O5 diminuent avec l'augmentation du numéro atomique de l'élément.
Comme le montre le tableau. 21, au sein du sous-groupe allant de l'azote au bismuth, les propriétés non métalliques diminuent et les propriétés métalliques augmentent. Dans l'antimoine, ces propriétés s'expriment également ; dans le bismuth, les propriétés métalliques prédominent ; dans l'azote, les propriétés non métalliques prédominent. Le phosphore, l'arsenic et l'antimoine forment plusieurs composés allotropiques.
Azote.
Reçu
En laboratoire, il peut être obtenu par réaction de décomposition du nitrite d'ammonium :
La réaction est exothermique, libérant 80 kcal (335 kJ), le récipient doit donc être refroidi pendant qu'elle se produit (bien que le nitrite d'ammonium doive être chauffé pour démarrer la réaction).
En pratique, cette réaction est réalisée en ajoutant goutte à goutte une solution saturée de nitrite de sodium à une solution saturée chauffée de sulfate d'ammonium, et le nitrite d'ammonium formé à la suite de la réaction d'échange se décompose instantanément.
Le gaz dégagé dans ce cas est contaminé par de l'ammoniac, de l'oxyde d'azote (I) et de l'oxygène, dont il est purifié par passage successif dans des solutions d'acide sulfurique, de sulfate de fer (II) et sur du cuivre chaud. L'azote est ensuite séché.
Une autre méthode de laboratoire pour produire de l'azote consiste à chauffer un mélange de bichromate de potassium et de sulfate d'ammonium (dans un rapport de 2:1 en poids). La réaction se déroule selon les équations :
L'azote le plus pur peut être obtenu par décomposition d'azotures métalliques :
L'azote dit « de l'air » ou « atmosphérique », c'est-à-dire un mélange d'azote avec des gaz rares, est obtenu en faisant réagir de l'air avec du coke chaud, ce qui produit le gaz dit « générateur » ou « air » - matière première pour la synthèse chimique et le carburant. Si nécessaire, l'azote peut en être séparé en absorbant le monoxyde de carbone.
L'azote moléculaire est produit industriellement par distillation fractionnée de l'air liquide. Cette méthode peut également être utilisée pour obtenir « l’azote atmosphérique ». Les installations et stations d'azote utilisant la méthode d'adsorption et de séparation des gaz par membrane sont également largement utilisées.
L'une des méthodes de laboratoire consiste à faire passer de l'ammoniac sur de l'oxyde de cuivre (II) à une température d'environ 700 °C :
L'ammoniac est extrait de sa solution saturée par chauffage. La quantité de CuO est 2 fois supérieure à celle calculée. Immédiatement avant utilisation, l'azote est purifié de l'oxygène et de l'ammoniac en passant sur du cuivre et son oxyde (II) (également ~700 °C), puis séché avec de l'acide sulfurique concentré et un alcali sec. Le processus est assez lent, mais il en vaut la peine : le gaz obtenu est très propre.
En figue. La figure 15.12 montre l'emplacement des cinq éléments du groupe V dans le tableau périodique. Tous sont des éléments p et la coque externe de leurs atomes est caractérisée par une configuration électronique. Dans le tableau 15.10 certaines propriétés des éléments du groupe V sont indiquées. À mesure que vous descendez vers le bas de ce groupe, les propriétés de ses éléments passent de non métalliques à métalliques. L'azote et le phosphore sont des non-métaux typiques. L'arsenic et l'antimoine présentent des signes de caractère métallique et le bismuth est un métal.
AZOTE ET PHOSPHORE
Ce sont les deux éléments les plus importants du groupe V.
Dans des conditions normales, l'azote est un gaz constitué de diatomique

Riz. 15.12. Position des éléments du groupe V dans le tableau périodique.
Tableau 15.10. Propriétés des éléments du groupe V

molécules Il est incolore, inodore et insipide et ne présente aucune réactivité notable.
Le phosphore existe sous trois formes allotropiques, dont les deux plus courantes sont le phosphore rouge et le phosphore blanc, le moins stable. La structure de ces allotropes a été discutée dans la Sect. 3.2. Dans le tableau 15.11, certaines propriétés du phosphore rouge et blanc sont comparées.
Bien que l’azote soit l’un des éléments les plus électronégatifs (voir tableau 2.2), sa réactivité est relativement faible. La raison en est que ses molécules diatomiques sont non polaires et que la triple liaison entre deux atomes
Tableau 15.11. Propriétés du phosphore blanc et rouge


Riz. 15.13. Quatre orbitales hybrides de l'atome d'azote.
L'azote qu'ils contiennent a une résistance très élevée. L'enthalpie de cette liaison est de + 944 kJ/mol.
L'azote et le phosphore peuvent pénétrer dans leurs composés avec des états d'oxydation de -3 à 4-5. Par exemple, dans l'ammoniac, l'azote a un état d'oxydation de +3 et dans l'ion nitrate, son état d'oxydation est de -1-5.
Les atomes de ces deux éléments sont capables d’attacher trois électrons, ce qui conduit à remplir leur sous-couche externe ; en conséquence, des ions nitrure et des ions phosphure se forment. Cependant, les deux éléments ont tendance à former des composés covalents. Dans ce cas, leurs atomes forment quatre orbitales hybrides, chacune ayant une orientation tétraédrique (Fig. 15.13). Trois de ces orbitales contiennent un électron. Ce sont ces électrons qui sont partagés lors de la formation de liaisons covalentes avec les électrons d'autres atomes. La quatrième orbitale contient une paire d'électrons non liants. Il peut être utilisé pour former un lien de coordination (donateur-accepteur). Par exemple, dans l’ion ammonium, le doublet isolé d’azote chevauche l’orbitale vacante de l’ion hydrogène.
ns 2 np 3.
AZOTE et ses composés
Dans la nature, on le trouve sous forme libre (teneur dans l'air - 78 % en volume), sous forme de minéraux (salpêtre), dans l'huile, dans les protéines végétales et animales.
Propriétés physiques
Dans des conditions normales, le N 2 est un gaz incolore, inodore et insipide.
kip. = -195,8°C.
Production d'azote :
a) dans l'industrie - liquéfaction de l'air,
b) en laboratoire : NH 4 NO 2 ® N 2 + H 2 O.
Propriétés chimiques de l'azote et de ses composés.
L'activité chimique de l'azote moléculaire est très faible, car grâce à la triple liaison covalente, la molécule est très forte (liaison E = 946 kJ/mol). Cependant, de nombreux composés différents sont connus pour l'azote, dans lesquels il présente les états d'oxydation -3, -2, -1, +1, +2, +3, +4, +5.
Composés azotés avec états d'oxydation négatifs.
3
1)NH 3 – ammoniac– un gaz incolore à l’odeur âcre, facilement liquéfiable.
Production d'ammoniac :
a) N 2 + 3H 2 « 2NH 3 (Kat : Fe, Al 2 O 3, K 2 O)
b) 2NH 4 CI + Ca(OH) 2 = CaCI 2 + 2NH 3 + 2H 2 O
Propriétés chimiques de l'ammoniac :
a) NH 3 + H 2 O « NH 4 OH (concentration maximale = 25 %)
b) NH 3 + HCI « NH 4 CI (mécanisme don.-acc. : NH 3 + H + = NH 4 +)
c) la combustion se produit différemment selon la quantité d'oxygène :
4NH 3 + 3O 2 (semaine) = 2N 2 + 6H 2 O
4NH 3 + 5О 2(g) = 2NO+ 6H 2 O
d) dans l'OVR, l'ammoniac est toujours un agent réducteur :
3CuO + 2NH 3 = 3Cu + N 2 + 3H 2 O
2) NH 4 OH – hydroxyde d'ammonium- base faible, instable, forme bien des composés complexes (ammoniac) :
Cu(OH) 2 + 4NH 4 OH = (OH) 2 + 4H 2 O
3) Sels d'ammonium :
a) les solutions dues à l'hydrolyse ont une réaction légèrement acide :
NH 4 CI + HON « NH 4 OH + HCI (NH 4 + + HON « NH 4 OH + H +)
b) la décomposition thermique des sels d'ammonium se déroule différemment selon la nature de l'anion : si l'anion est un oxydant, alors la réaction de décomposition est irréversible :
NH 4 CI « NH 3 + HCI
NH 4 NO 3 = N 2 O + 2H 2 O
4) Amides se forment lorsque les atomes d'hydrogène de l'ammoniac sont remplacés par du métal :
2K + 2NH 3 = 2KNH 2 + H 2
5) Nitrures connus pour les métaux et les non-métaux et, selon leur nature, se décomposent différemment :
a)Li 3 N + 3H 2 O = 3LiOH + NH 3
Ca 3 N 2 + 6HCI = 3CaCI 2 + 2NH 3
b)CI 3 N + 6HCI = 3HCIO + NH 3
État d'oxydation - 2
N 2 H 4 – hydrazine(diamide) est un liquide incolore, un bon solvant, moins stable que l'ammoniac.
Préparation de l'hydrazine :
2NH 3 + NaCIO = N 2 H 4 + NaCI + H 2 O
Propriétés chimiques de l'hydrazine :
N 2 H 4 + H 2 O « + +OH -
H 2 O « 2+ +OH -
b) Brûle en azote : N 2 H 4 + O 2 = N 2 + 2H 2 O
c) Agent réducteur fort : N 2 H 4 + 2H 2 O 2 = N 2 + 4H 2 O
État d'oxydation - 1
NH 2 OH – hydroxylamine, une substance cristalline blanche, occupe une position intermédiaire entre H 2 O 2 et l'hydrazine N 2 H 4.
Préparation de l'hydroxylamine :
HNO 3 + 6H = NH 2 OH + 2H 2 O
Propriétés chimiques de l'hydroxylamine :
a) Solutions aqueuses – bases faibles :
NH 2 OH + H 2 O « + +OH -
b) Les sels d'hydroxylammonium (stables) sont donnés avec des acides :
NH 2 OH + HCI = + + CI -
c) Se décompose (réaction de disproportion) :
3NH 2 OH = NH 3 + N 2 + 3H 2 O
d) Comme H 2 O 2, selon l'environnement il présente une dualité rédox :
d'accord: 2NH 2 OH + 4FeSO 4 + 3H 2 SO 4 = 2Fe 2 (SO 4) 3 + (NH 4) 2 SO 4 + 2H 2 O
restauré : 2NH 2 OH + I 2 + 2KOH = N 2 + 2KI + 4H 2 O
Les composés azotés les plus importants avec s positif. O.
État d'oxydation + 3
1) N2O3- le monoxyde d'azote (III) ou « anhydride nitreux » est un liquide bleu-vert instable :
a) N 2 O 3 ® NO 2 (gaz brun avec une odeur âcre) + NO (gaz incolore) ;
b) forme un acide instable avec l'eau : N 2 O 3 + H 2 O « 2 HNO 2
2) HNO2– acide nitreux (force moyenne) :
HNO 2 « H + + NO 2 - (K = 5,1 . 10 -4).
3) Nitrites– les sels de l'acide nitreux, comme les autres composés N +3 dans les réactions redox, présentent une dualité :
d'accord: KNO 2 + NH 3 = N 2 + H 2 O
restauré : KNO 2 + H 2 O 2 = KNO 3 + H 2 O
État d'oxydation maximal + 5
1) N2O5- l'oxyde nitrique (V) ou « anhydride nitrique » est une substance cristalline incolore ; Dissous dans l'eau, il donne de l'acide.
2) HNO3– acide nitrique, fort : HNO 3 ® H + + NO 3 - .
3) Nitrates– les sels d'acide nitrique :
a) en laboratoire, vous pouvez obtenir du N 2 O par la réaction de décomposition thermique : NH 4 NO 3 ® N 2 O + H 2 O ;
b) les nitrates métalliques, selon la position du métal dans la série d'activités, se décomposent différemment :
à gauche Mg : nitrate ® nitrite + O 2
Mg-Cu : nitrate ® oxyde métallique + NO 2 + O 2
à droite Cu : nitrate ® métal + NO 2 + O 2 .
4) tous les composés N +5 sont des agents oxydants forts ; interaction de l'acide nitrique avec les métaux :
a) aucun métal ne libère d'hydrogène à partir de HNO 3 ;
b) à la normale par rapport aux métaux Al, Zn, Cr, Fe, Pb, Au, Pt sont passivés ;
c) lorsqu'il est chauffé, il interagit avec la plupart des métaux, et plus le métal est actif et plus l'acide est dilué, plus la réduction de N +5 est forte :
inactif Meh: Cu + 4HNO 3 (conc.) = Cu (NO 3) 2 + 2NO 2 + 2H 2 O
3Cu + 8HNO 3(dil.) = 3Cu(NO 3) 2 + 2NO + 4H 2 O
actifs. Meh: 4Ca + 10HNO3(conc.) = 4Ca(NO3)2 + N2O + 5H2O
5Ca + 12HNO3 (dilué) = 5Ca(NO3)2 + N2 + 6H2O
4Ca + 9HNO 3 (ultra dil.) = 4Ca(NO 3) 2 + NH 3 (ou NH 4 NO 3) + 3H 2 O
PHOSPHORE et ses composés
On le trouve dans la nature : a) sous forme de minéraux (phosphorites et apatites), b) dans les protéines végétales et animales, c) dans les tissus cérébraux et les os.
Propriétés physiques
Allotropène :
UN) phosphore blanc(P 4) – structure tétraédrique, toxique, brille dans le noir, soluble dans le sulfure de carbone ; pendant le stockage, il se transforme progressivement en phosphore rouge, il est donc stocké sous l'eau ;
b) phosphore rouge(P 2 n – polymère) – il existe plusieurs formes de structure, non toxiques, insolubles dans le sulfure de carbone, stables au stockage, moins réactives ;
V) phosphore noir– structure pyramidale, en couches, semblable au graphite, semi-conducteur.
Obtention du phosphore :
Le minéral phosphorite est calciné avec du coke et du sable :
Ca 3 (PO 4) + 5C + 3 SiO 2 = 3CaSiO 3 +2P +5CO
Propriétés chimiques du phosphore et de ses composés.
L'activité chimique du phosphore varie en fonction de sa modification allotropique. Principaux états d'oxydation : -3, +1, +3, +5.
1) Réactions du phosphore avec des substances simples :
propriétés réparatrices : a)P + O 2 ® P 2 O 3, P 2 O 5;
b) P + CI 2 ® PCI 3, PCI 5 ; c) 2P + 3S ® P 2 S 3
propriétés oxydantes : a) 2P + 3H 2 ® 2РН 3, b) 2P + 3Ca ® Ca 3 P 2
2) Réactions du phosphore avec des substances complexes :
a) la réaction avec l'eau ne se produit pas,
b) 2P + 5H 2 SO 4 (conc.) ® 2H 3 PO 4 + 5SO 2 + 2H 2 O,
c) P + HNO 3 (conc.) ® H 3 PO 4 + NO (ou NO 2).
3)État d'oxydation minimum - 3
UN) PH3– la phosphine, un gaz toxique à l'odeur d'ail,
allumé : 2РH 3 + 4О 2 = Р 2 О 5 + 3H 2 O,
avec certains acides il donne des sels de phosphonium : РH 3 + HI = РH 4 I
b) phosphures métalliques– composés ionocovalents
Mg 3 P 2 + 6 H 2 O = Mg(OH) 2 + 2РН 3,
Ca 3 P 2 + 6HCI = CaCI 2 + 2PH 3.
4)État d'oxydation + 1
H 3 PO 2 ou H[PO 2 H 2 ] – acide hypophosphoreux, fort ; les sels sont des hypophosphites, très solubles, de bons agents réducteurs.
5)État d'oxydation + 3
a) P 2 O 3 – anhydride de phosphore, a plusieurs modifications (P 2 O 3) n, cristaux toxiques incolores ;
b) H 3 PO 3 ou H 2 [PO 3 H] – acide phosphoreux, force moyenne ;
c) les sels de phosphite, solubles uniquement dans les métaux alcalins (K 2 [PO 3 H]),
PCI 3 + CI 2 = PCI 5,
PCI 3 + O 2 = PCI 3,
PCI 3 + HCI ® N.
6)État d'oxydation maximal +5
a) P 2 O 5 – anhydride de phosphore, poudre blanche, a une modification (P 2 O 5) 2, un puissant agent déshydratant : P 2 O 5 + 2H 2 O = 2HPO 3 ;
b) HPO 3 - méta, H 4 P 2 O 7 - pyro, H 3 PO 4 - acide orthophosphorique, force moyenne ;
c) les sels méta-, pyro-, ortho-phosphate ;
d) les halogénures sont assez actifs :
PCI 5 + 4H 2 O = H 3 PO 4 + 5HCI,
RF 5 + HF = N.
CARACTÉRISTIQUES GÉNÉRALES DU SOUS-GROUPE
7 N, 15 P, 33 As, 51 Sb, 83 Bi. Dans le sous-groupe de haut en bas, les propriétés métalliques augmentent naturellement, ce qui est cohérent avec les valeurs des états d'oxydation présentés par les éléments des composés, ainsi qu'avec certaines propriétés physiques. Bien que l'allotropie de P, As et Sb crée certaines difficultés lors de la combinaison des propriétés physiques dans un seul tableau :
Tableau 11
Propriétés des éléments du cinquième groupe, le sous-groupe principal
Propriétés chimiques
1. Avec des substances simples, ils donnent une variété de composés, présentant différentes natures rédox :
a) propriétés réparatrices
2P + 3Cl 2 = 2PCl 3 (ou PCl 5) ;
4As + 5O 2 = 2As 2 O 5 ;
2Bi + 3Cl 2 = 2BiCl 3.
b) Propriétés oxydantes
2P + 3Ca = Ca3P2 ;
2As + 3Mg = Mg3As 2.
2. Ils réagissent avec les acides différemment selon la nature de l'élément du sous-groupe :
2P + 5H 2 SO 4 = 2H 3 PO 4 + 5SO 2 + 2H 2 O ;
3As + 5HNO 3 + 2H 2 O = 3H 3 AsO 4 + 5NO ;
3Sb + 5HNO 3 = 3HSbO 3 + 5NO + H 2 O ;
2Sb + 6H 2 SO 4 conc. = Sb 2 (SO 4) 3 + 3SO 2 + 6H 2 O ;
Bi + 4HNO 3 = Bi(NO 3) 3 + NON + 2H 2 O.
Il faut rappeler que les éléments du sous-groupe arsenic apparaissent après l'hydrogène dans la série d'activités.
3. L'hydrolyse des composés se produit de différentes manières :
a) PCl 3 + 3H 2 O = H 3 PO 3 + 3HCl ;
BiCl 3 + H 2 O ® BiOCl ¯ + 2HCl.
(SbCl 3) (SbOCl)
b) NaNO 2 + H 2 O Û HNO 2 + NaOH ;
K 3 PO 4 + H 2 O Û K 2 HPO 4 + KOH;
K 3 AsO 4 + H 2 O Û K 2 HAsO 4 + KOH.
4. Les modifications des propriétés acido-basiques des éléments se manifestent de différentes manières
Tableau 12
Les composés les plus importants d'éléments du cinquième sous-groupe
5. La capacité rédox des composés dépend du degré d'oxydation de l'élément :
UN) E-3- les agents réducteurs
2NH 3 + 6KМnO 4 + 6KOH = 6K 2 MnO 4 + N 2 + 6H 2 O ;
2PH 3 + 4O 2 = P 2 O 5 + 3H 2 O.
agents oxydants:
2KNO 2 + 4H 2 S = 3S + N 2 + K 2 S + 4H 2 O
E +3 2BiCl 3 + 3SnCl 2 + 18KOH = 2Bi + 3K 2 SnO 3 + 12KCl + 9H 2 O.
les agents réducteurs:
10KNO 2 +4KМnO 4 +11H 2 SO 4 =10HNO 3 +4MnSO 4 +7K 2 SO 4 +6H 2 O
V) E +5 – agents oxydants
Na 3 SbO 4 + 2NaJ + H 2 SO 4 = Na 3 SbO 3 + J 2 + Na 2 SO 4 + H 2 O.
Éléments du groupe 6. (Chalcogènes)
La structure de la coque électronique : ... ns 2 np 4.
8 O, 16 S, 34 Se, 52 Te, 84 Po (radioactif).
OXYGÈNE et ses composés
L'élément le plus répandu sur Terre : 21 % (en volume) dans l'air, une partie de H 2 O, des minéraux et des substances organiques.
Propriétés physiques
Existe sous forme de substances :
a) « oxygène » O 2 – un gaz sans couleur, sans goût, sans odeur, non toxique ; t° bouillir. = -183°C; ;
b) « ozone » O 3 – un gaz bleu avec une odeur âcre, toxique, un oxydant très puissant ; t° bouillir. = -111,8°C.
Propriétés chimiques
1. Dans une seule réaction, l’oxygène est un agent réducteur :
О 2 + 2F 2 = 2OF 2 ().
2. Dans tous les autres cas, l'oxygène est un agent oxydant, surtout s'il est à l'état atomique :
a) Oxydation de substances simples
2H 2 + O 2 = 2H 2 O;
S + O 2 = SO 2;
2Ca + O2 = 2CaO.
b) Oxydation de substances complexes
R 2 O 3 + O 2 = R 2 O 5;
2SO 2 + O 2 = 2SO 3 ;
2PCl 3 + O 2 = 2POCl 3.
c) Combustion de substances inorganiques complexes
2H 2 S + O 2 insuffisant = 2S + 2H 2 O;
2H 2 S + 3O 2 g. = 2SO 2 + 2H 2 O.
d) Combustion de substances organiques
CH 4 + 2O 2 = CO 2 + 2H 2 O ;
C 2 H 5 OH + 3O 2 = 2CO 2 + 3H 2 O.
SOUFRE et ses composés
Trouvé dans la nature sous sa forme native ; sous forme de minéraux (sulfures, sulfates) ; dans l'huile; en protéines.
Propriétés physiques
Dur, cassant, jaune, insoluble dans H 2 O, mais soluble dans le sulfure de carbone et l'aniline. Allotropique : soufre rhombique (forme a) ; monoclinique (b - soufre); plastique, etc. La modification la plus stable est a (t° pl. » 113°C), on lui attribue la composition S 8.
Propriétés chimiques
Les composés, présentant différents degrés d'oxydation, ont des capacités rédox différentes.
Tableau 13
Les composés soufrés les plus importants
1. Le soufre, interagissant avec des composés complexes, modifie son état d'oxydation de différentes manières :
a) S + 2H 2 SO 4 conc. = 3SO 2 + 2H 2 O ();
b) S + 6HNO 3 conc. = H 2 SO 4 + 6NO 2 + 2H 2 O ();
c) 3S + 6NaOH = 2Na2S + Na2SO3 + 3H2O
réaction de disproportion
2. Connexions S-2
UN) H2S– le sulfure d'hydrogène, gaz toxique à l'odeur d'œuf pourri, puissant réducteur : 2H 2 S + O 2 = 2S + 2H 2 O ;
H 2 S + Cl 2 = 2HCl + S.
b) Solution H 2 S – acide faible
H 2 S Û N + + HS - (K 1 = 6 × 10 -8).
c) Sels – sulfures ; les sels de métaux alcalins et alcalino-terreux sont très solubles et s'hydrolysent ; les sulfures de métaux p et d sont pratiquement insolubles.
G) Sulfures exposition de métaux principal nature:
K 2 S + H 2 O Û KHS + KOH;
Exposition de sulfures non métalliques acide nature:
SiS 2 + 3H 2 O = H 2 SiO 3 + 2H 2 S.
d) Thiosols: semblable aux sels ordinaires dans lesquels les atomes O sont remplacés par S :
Na 2 S + CS 2 = Na 2 CS 3 – thiocarbonate de sodium ;
Na 2 CS 3 + 2HCl = 2NaCl + H 2 CS 3
instable
e) Polysulfures: le soufre, comme l'oxygène dans les peroxydes, peut former des sels : solution Na 2 S + ´S = Na 2 S n - polysulfure de sodium (contient des ponts sulfure - S - S - S - S -).
3. Connexions S+4
a) SO 2 – dioxyde de soufre avec une odeur âcre, toxique ;
b) Solution SO 2 - acide sulfureux H 2 SO 3, force moyenne
H 2 SO 3 Û H + + HSO 3 - (K 1 = 1,3 × 10 -2) ;
c) Sels – sulfites, bien hydrolysés
Na 2 SO 3 + H 2 O Û NaHSO 3 + NaOH.
d) Dans les réactions redox, tous ces composés présentent une dualité :
SO 2 + 2H 2 S = 3S + 2H 2 O;
2SO 2 + O 2 = 2SO 3.
e) chlorure de thionyle : SOCl 2
SOCl 2 + 2H 2 O = H 2 SO 3 + 2HCl.
4. Connexions S+6
UN) SỐ 3– l'anhydride sulfurique ; agent oxydant allotropique et puissant. Réagit très vigoureusement avec l'eau
SO 3 + H 2 O ® H 2 SO 4 + 89,1 kJ/mol;
b) H2SO4– l'acide sulfurique, liquide huileux incolore ; fort.
H 2 SO 4 conc. – agent oxydant très puissant ; papier carbonisé, sucre, bois :
C + 2H 2 SO 4 conc. = CO 2 + 2SO 2 + 2H 2 O.
Individuellement, son comportement avec les métaux :
aux températures ordinaires, il n'interagit pas avec de nombreux métaux (« passive » Cr, Fe, Al, Zn, ...) ;
à des niveaux élevés, il réagit avec presque tous les métaux à l'exception de l'Au et du Pt.
C'est un très bon agent déshydratant car... avec l'eau forme des hydrates H 2 SO 4 × nH 2 O :
V) Sels: moyen – K 2 SO 4 (sulfates) ;
acide – KHSO 4 (sulfates d'hydrogène);
vitriol – CuSO 4 × 5H 2 O ; ZnSO 4 × 7H 2 O;
alun – sels doubles, hydrates cristallins
KAl(SO 4) 2 × 12H 2 O; NH 4 Fe(SO 4) 2 × 12H 2 O.
G) Oléum– une solution de SO 3 dans H 2 SO 4, alors que la réaction se produit partiellement
2SO 3 + H 2 O = H 2 S 2 O 7 acide pyrosulfurique.
d) Chlorure de sulfuryle SO2Cl2
SO 2 Cl 2 + 2H 2 O = H 2 SO 4 + 2HCl.
e) Acides peroxosulfuriques– contiennent des ponts peroxydes
H 2 SO 5 – peroxomonosulfure,
H 2 S 2 O 8 – peroxodisulfure ;
agents oxydants puissants.
et) Thiosulfates– contiennent du soufre dans différents états d'oxydation : S +6 et S -2 ; en raison de la présence de S-2, les composés sont de puissants agents réducteurs.
Na 2 S 2 O 3 + Br 2 + H 2 O = 2HBr + S + Na 2 SO 4.
L'acide correspondant H 2 S 2 O 3 (thiosulfurique) est instable :
Na 2 S 2 O 3 + H 2 SO 4 = Na 2 SO 4 + H 2 S 2 O 3.
Éléments du groupe 7a. (Halogènes )
La structure de la coque électronique : ... ns 2 np 5.
9 F, 17 Cl, 35 Br, 53 J, 85 At (radioactif). À l’état libre, ils existent sous forme de molécules diatomiques.
Propriétés physiques
Toxique, avec une odeur âcre F 2 – gaz jaune pâle, point d'ébullition. = -188,2 °C ;
Cl 2 – gaz vert-jaune ; t° bouillir. = -34,1°C;
Br 2 – liquide rouge ; t° bouillir. = 59,2°C;
J 2 – cristaux violets ; t° bouillir. = +185,5°C.
Propriétés chimiques
1) Le plus actif est F 2, même le verre et l'eau brûlent dans son atmosphère :
2F 2 + SiO 2 = SiF 4 + O 2;
2F 2 + 2H 2 O = 4HF + O 2.
2) Les halogènes en amont (en tant qu'agents oxydants plus puissants) remplacent ceux en aval de divers composés :
F 2 + 2KCl (KBr, KJ) = 2KF + Cl 2 (Br 2, J 2).
3) Lorsque les halogènes sont dissous dans des solutions aqueuses et alcalines, des réactions de dismutation se produisent ; En fonction de la température, différents produits de réaction sont obtenus :
Cl 2 + H 2 O = HCl + HClO;
Cl 2 + 2NaOH = NaCl + NaClO;
4) Halogénures d'hydrogène– ce sont tous des gaz incolores (HF, du fait des liaisons hydrogène, forme des associés H x F x).
Dans la série : HF – HCl – HBr – HJ, la force d’adhésion diminue et les propriétés réductrices augmentent :
MnO 2 + 4HCl = Cl 2 + MnCl 2 + 2H 2 O ().
5) Lorsqu'ils sont dissous dans l'eau, des acides forts sont obtenus ; L'HJ est le plus fort (à l'exception de l'acide fluorhydrique faible :
H 2 F 2 Û H + + HF 2 -)
6) Halogènes les éléments ont un caractère acido-basique différent, qui se manifeste :
a) dans les réactions d'hydrolyse
AlCl 3 + H 2 O Û AlOHCl 2 + HCl;
PBr 3 + 3H 2 O = H 3 PO 3 + 3HBr.
b) dans les réactions de formation complexes
2NaF + SiF 4 ® Na 2 ;
pour un certain nombre de ligands F - ; Cl- ; Br - ; J - la stabilité des complexes diminue.
7) Halogènes dans des états d'oxydation plus élevés
a) oxydes : Cl 2 O ; (ClO2)2; (ClO3)2; Cl207;
b) acides : HClO ; HClO2 ; HClO3; HClO4 ;
pour un certain nombre d'acides ®, la force augmente, pour les acides et leurs sels de la même série, les propriétés oxydantes diminuent.
c) les acides et leurs sels se décomposent lorsqu'ils sont chauffés :
d) composés entre halogènes : beaucoup d’entre eux sont connus ; La composition correspond à la position des éléments dans le tableau périodique (des éléments plus électronégatifs apparaissent à la fin des formules) :
ClF, BrF, JF ; ClF3, JF3; ClF5, ClOF3; BrF 7, JO 3 F, etc.;
sont de nature acide : JF 7 + 4H 2 O = 7HF + HJO 4 ;
ClO 3 F + 2NaOH = NaClO 4 + NaF + H 2 O.
Questions pour consolider le matériel :
1. Quelle substance se forme lorsque l'arsenic réagit avec l'acide nitrique concentré - a)Аs(NO3)3 ? b)H 3 AsO 4 ? c) Comme 2 O 3 ?
2.Quelle substance réagit avec un acide concentré pour libérer du chlore libre – a)CaC1 2 ? b)MnO2 ? c) CrC1 3 ?
Nous recommandons également
 Butin de guerre : comment les soldats soviétiques ont « volé » la population allemande
Butin de guerre : comment les soldats soviétiques ont « volé » la population allemande
 Robert Galbraith. Au service du mal. Au service du mal Robert Galbraith au service du mal mobi
Robert Galbraith. Au service du mal. Au service du mal Robert Galbraith au service du mal mobi
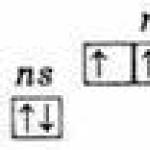 Éléments P du groupe V Valence maximale des éléments du groupe 5 du sous-groupe principal
Éléments P du groupe V Valence maximale des éléments du groupe 5 du sous-groupe principal
 Histoires de vie, de mort et de neurochirurgie
Histoires de vie, de mort et de neurochirurgie
 Concepts et principaux types d'opérations bancaires modernes
Concepts et principaux types d'opérations bancaires modernes
 Pauvre Lisa, télécharge fb2. Pauvre Lisa. Nikolaï Karamzine
Pauvre Lisa, télécharge fb2. Pauvre Lisa. Nikolaï Karamzine